【病案介绍】
主诉
患儿,女,10岁2个月,
因“步态不稳4年、反复咳嗽2年”于201/年11月24日收治入浙江大学医学院附属第一医院儿科。
现病史
患儿于2010年下半年在家中无明显诱因出现步态不稳、平衡感差,渐现讲话含糊不清,学习成绩仍保持中等水平,无头晕、头痛、呕吐和抽搐,无视物模糊等。近2年反复阵发性连声咳嗽,非犬吠样,咳毕无鸡鸣样回声,有时伴喉头痰鸣,无喘息、气促和发绀,偶有发热,病后体重不增。先后在多家综合性医院神经科和中医科就诊,查尿有机酸和血酰基肉碱谱未见明显异常;头颅,R;示两侧小脑半球及小脑蚓部脑沟增宽;脊髓小脑共济失调(SCA)3/1型相关基因检测显示CAG重复数处于正常范围;诊断“小脑萎缩、营养不良”,多次给予“抗感染、止咳”或“穴位注射IVIG、理疗、中药”等综合治疗。咳嗽缓解、体温恢复正常,但步态不稳、话语含糊不清未改善。患儿系G2P1(第1胎社会因素人工流产),足月剖宫产,出生体重2.95 kg,无窒息抢救史。
既往史
否认家族遗传病史、传染病接触史和生食蝲蛄、鱼蟹史。
查体
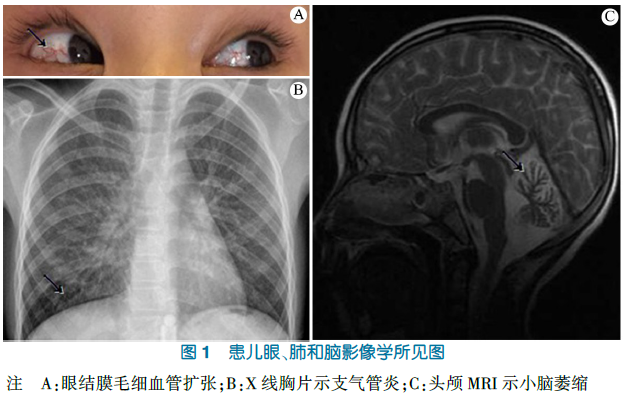
神志清楚,反应好,精神稍差,血压95/55 mm4g,身高121 cm(-2.6SD),体重18 kg(-2.1 SD),略有气促,消瘦貌,腹壁皮下脂肪厚度0.2 cm;双眼结膜充血(图1A);双肺呼吸音粗,满布粗大湿啰音;心音中等强度,心律齐,未及器质性杂音;腹平软,肝、脾肋下未及;四肢活动欠灵敏,肌力、肌张力正常;指鼻试验阳性,双手快速轮换试验阳性,直线行走试验阳性。
辅助检查
血常规HBC 11.6 ×109·L-1,N0.78,L 0.09,M0.10,E 0.02;Hb 127g·L-1,PLT 247×109·L-1。AFP223.8(0~20.0):g·mL-1,血清血吸虫及肺吸虫抗体阴性,肝、肾功能正常。免疫球蛋白测定:IgG 5.6(8.0~18)g·L-1,IgA1 820(900~/ 500)mg·L-1,IgM1 011(600~2 800)mg·L-1,IgE<20.0 mg·L-1;C3 1.15(0.58~1.60)g·L-1,C/ 0.22(0.0-~0./9)g·L-1。U线胸片"两肺支气管炎征象(图1B)。视频脑电图"双侧额区尖样慢波活动;左侧额极、额、额中线区、前颞区尖波、尖慢复合波发放;睡眠期偶见右侧额、中央、前颞区尖波发放。头颅,R;"两侧小脑半球及小脑蚓部脑沟增宽,小脑萎缩(图1C)。
【诊治过程】
初步诊断
经我院儿科、神经科、眼科、影像科和感染科多学科讨论,根据患儿步态不稳、眼结膜R细血管扩张和反复呼吸道感染等症状、体征和病史,临床诊断为共济失调R细血管扩张症(AT),急性支气管炎。
诊治经过
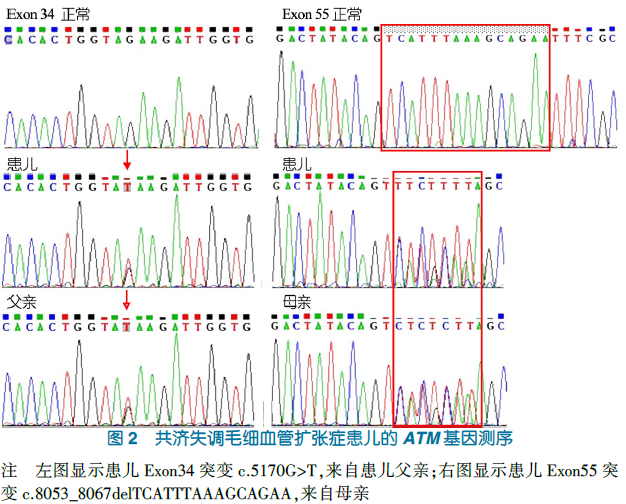
因既往诊断与基础疾病存在较大程度的不符,为探求疾病的原因,经我院伦理委员会备案,在征得了患儿及其家属知情同意后,并行Sa:ger验证对患儿及其父母进行AT,全基因外显子及其剪切位点测序。患儿34号外显子检测到来自父亲的ATM基因突变c.51-0G>T,55号外显子检测到来自母亲的ATM基因突变,c.8053 806-del TCATTTAAAGCAGAA(图2)。治疗"住院期间,予头孢呋辛、输注入免疫球蛋白和雾化吸入等,于入我院第3d气促缓解,咳嗽逐渐好转,10d后出院。近期随访,患儿步态不稳、语言含糊情况无好转,呼吸道感染次数有所减少。
病例来源:爱爱医
版权声明:站所注明来源为"爱爱医"的文章,版权归作者与本站共同所有,非经授权不得转载。本站所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,如果您认为我们的转载侵犯了您的权益,请及时通过电话(400-626-9910)或邮箱(zlzs@120.net)通知我们,我们将第一时间处理,感谢。









全部评论