【病案介绍】
主诉
女,36 岁。
男,30 岁。
因“反复眼黄、尿黄、皮肤黄36 年”于2016 年6 月15 日入院。
因“眼黄、尿黄、皮肤黄7年”于2016 年5月30日就诊我院。
现病史
缘于出生后即被发现眼黄、皮肤黄,尿黄,饮食等日常生活如常,无皮肤瘙痒。生长发育正常,尿黄因过度劳累或熬夜后加重,经休息后缓解。多次在多家医院就诊,检查提示总胆红素(TBIL)波动于120~150μmol/L,谷丙转氨酶(ALT)正常,未明确诊断,常规治疗无好转。10 年前,妊娠时血清TBIL升高至250μmol/L,分娩后恢复至以前水平。
缘于2009 年春无明显诱因下出现眼黄、尿黄、皮肤黄,血清TBIL 最高时达75.0μmol/L,尿黄可因过度劳累、饮酒或熬夜后加深,经休息后减轻。就诊我院时TBIL 64.3 μmol/L,IBIL 45.2 μmol/L,MRCP、腹部彩超提示:脾肿大(厚约62 mm),其他发病情况类似例1。
查体
巩膜、皮肤中度黄染,未见肝脏、蜘蛛痣,腹平坦、肝脾未触及,肝区无叩痛。
辅助检查
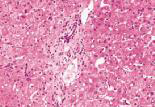
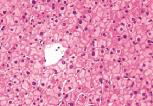
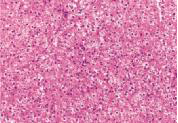
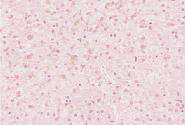
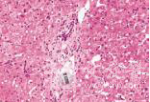
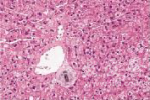
化验:TBIL 153.9μmol/L,IBIL67.0 μmol/L,血清HBsAg、HBV DNA、抗-HCV、HCV RNA、抗-HIV、抗HAV-IgM、抗HEV-IgM、抗CMV-IgM、抗EBV-IgM、常见自身抗体均阴性。骨髓穿刺检查、血常规、尿常规、血清甲胎蛋白、凝血功能和其他生化指标均无明显异常。上腹部CT检查无明显异常。肝穿刺活组织检查标本送广州金域医学检验中心病理科检查,发现肝小叶结构存在,部分汇管区轻微扩大,未见炎性细胞浸润。区域性毛细胆管轻度扩张,淤胆性色素颗粒有围绕毛细胆管现象(图1~3),将外周血送往北京宏微特斯生物科技有限公司行基因检测,发现患者尿苷葡萄糖醛酸转移酶基因突变点位于启动子上游PBREM-3263(-3279)以编码区外显子EX0N1 上的GGAAGA Gly71Arg,最终确诊为Gilbert综合征。
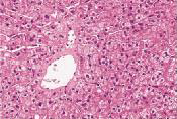
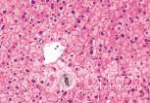
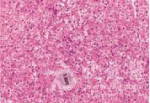
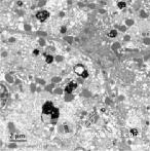
肝穿刺组织病理学检查发现肝小叶结构存在,未见炎性反应,中央静脉周围肝细胞内少量淤胆性色素颗粒,普鲁士蓝染色可见Kupffer 细胞内少许铁颗粒,肝细胞内可见多少不一的淤胆性色素颗粒,区域性毛细胆管扩张(图4~7)。基因检测发现患者基因突变点位于启动子上游PBREM-3263。最终确诊为Gilbert综合征。
病例来源:爱爱医
版权声明:站所注明来源为"爱爱医"的文章,版权归作者与本站共同所有,非经授权不得转载。本站所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,如果您认为我们的转载侵犯了您的权益,请及时通过电话(400-626-9910)或邮箱(zlzs@120.net)通知我们,我们将第一时间处理,感谢。











全部评论