【病案介绍】
主诉
女,14岁
男,10月龄。
2014年5月7日因“发现蛋白尿1月余”就诊于复旦大学附属儿科医院(我院),以蛋白尿
原因待查收入我院。
2015年4月因“心脏杂音,肌张力低下”就诊并收入我院。
现病史
患儿1个月前因“白头发增多”就诊当地医院,查尿常规蛋白3+,未见全身水肿,无尿频、尿急和尿痛,无肉眼血尿及少尿,SCr 98.1 umol·L-1,BUN 6.1 mmol·L-1,内生肌酐清
除率74 mL·min-1·S-1,肾脏病理学检查提示“早期硬化肾小球肾炎”,予黄葵胶囊和贝那普利口服,尿蛋白仍3+。
患儿出生后因“呕吐14 h”入住当地医院,诊断“新生儿不全性肠梗阻、败血症、先天性心脏病、新生儿肺炎、新生儿窒息、新生儿脑病、肝功能异常、鞘膜积液和足月小样儿”,经治疗好转出院(具体治疗不详)。1个月前因“右侧腹股沟复发性包块8个月”再就诊当地医院,查体发现生长发育落后、左眼睑下垂和四肢肌张力低下;头颅MR]示双侧大脑萎缩,行右侧腹股沟嵌顿性斜疝复位和疝囊高位结扎术。患儿因精神、运动发育落后来我院就诊,心脏超声示:肺动脉狭窄、动脉导管未闭、房间隔缺损、降主动脉流速增快、永存左上腔静脉。
既往史
既往体健,否认食物、药物过敏史。
个人史
患儿系G:P:,足月顺产,出生体重3 400 g,
患儿系G,P,,足月顺产,出生体重1 700 g,血性羊水,轻度窒息。生长发育迟缓,目前不能抬头、不能坐;
查体
血压110/70 mmHg,身高152.0 cm(P50),体重44.0 kg(P50),神志清楚,精神可。全身未见水肿。心、肺、腹部和神经系统查体未见异常。
血压88/50 mmHg,身高65.0 cm(P50),体重5.o kg(P50),神志清楚,精神、反应欠佳,左侧上眼睑下垂,全身未见明显水肿。HR 130·min-1,律齐,心前区可闻及收缩期m/6杂音,以胸骨左缘第2、3肋间最为明显;腹软,无压痛及反跳痛;四肢肌张力低下,不能竖头,不能坐立,克氏、布氏征均阴性。
辅助检查
尿沉渣:蛋白2+、RBC 0.41·HP-1、WBC 0.29·HP-1。;尿蛋白/CR 2.79,尿钙/CR0.04,24 h尿蛋白2.87 g。尿微量蛋白A1M 6.12 mg·L-1,A1MU/CR22.0 mg·g-1,ALBU 741.0 mg·L-1,ALBU/CR 2 667mg·g-1。,IGGU 22.7 mg·L-1,IGGU/CR 81.7 mg·g-1,NAG 5.2 U·L-1,NAG/CR 2.12 U·mol-1。,尿转铁蛋白31.9 mg·L-1。血常规、肝功能、血电解质、血脂、免疫球蛋白和补体指标均正常。自身抗体阴性。SCr 74.0umol·L-1,BUN 6.6 mmol·L-1,尿酸44umol.L-1,甲状旁腺素182 Pg·mL-1。血气分析:pH 7.270、BE一4.2 mmol·L-1。
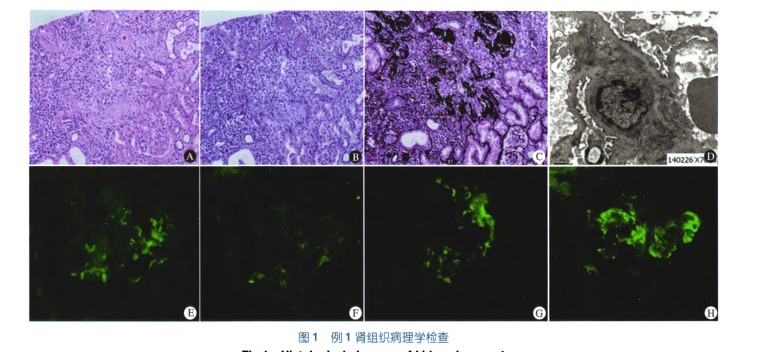
调阅患儿外院的肾活检病理切片,我院行重新阅片:光镜12个肾小球,7个球性硬化,1个局灶阶段性肾小球硬化,4个肾小球正常,明显肾小管萎缩及间质纤维化,肾血管无异常(图1A—c);电镜下足细胞足突部分融合(图1D);免疫荧光显微镜显示(图lE—H):系膜区有IgM、IgA、IgG、Clq和C3沉积(+—++)。DTPA示双肾灌注欠佳,功能受损,排泄延迟,双肾。肾小球滤过率(GFR,未标化)48.4 mL·min。(左肾26.5、右肾21.9),肾脏MR示左肾实质多发性异常信号,髓质海绵肾待排除,右肾偏小伴右肾可疑异常信号。肾脏磁共振水成像(MRU)和膀胱逆行造影(MCU)未见异常。
尿沉渣:蛋白3+、RBC 0—1·HP-1,WBC2~3·HP-1;24 h尿蛋白1.04 g;血常规:Hb 85 g·L-1,RBC 2.64×10“·L-1;血生化:ALT 24 u·L-1,AST 43U·L-1,总蛋白44.8 g·L-1。,白蛋白22.5 g·L-1,总胆固醇5.43 mmol·L-1,三酰甘油2.38 mmo]·L-1,SCr 14.0I-Lmol·L-1,BUN 4.2 mmol·L-1,尿酸218.0umol·L-1。
血电解质正常。TORCH抗体均阴性,梅毒、HIV和乙肝血清学检查均阴性。免疫球蛋白和补体均正常。血气分析:pH 7.496,BE一0.5 mmol·L-1。泌尿系统B超检查未有异常发现。

【诊治过程】
初步诊断
局灶阶段性肾小球硬化(FSGS)、慢性肾脏病2 期。
先天性心脏病,肾病综合征,先天性脑发育不良、左侧上眼睑下垂。
诊治经过
因家长拒绝糖皮质激素治疗,继续予贝那普利和和黄葵胶囊13服治疗。
行心导管造影和经皮动脉导管未闭封堵术,术后予阿司匹林、**和螺内酯口服。
【其他】
【家族史】
患儿父母体健,非近亲婚配,母亲妊娠史2旬旬-2,患儿有一个哥哥,体健。
父母亲体健,非近亲婚配,母亲妊娠史1-0旬-1,家族成员否认遗传性疾病史。
【讨论】
测序分析
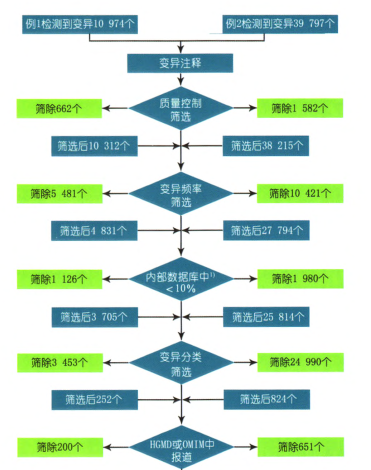
高通量测序: 使用Hiseq 2500高通量模式行PEl00测序,按Hiseq 2500标准流程进行。通过Q30的标准对原始数据进行数据过滤得到cleandata和高质量的测序结果。使用bwa比对软件,使用samtool和pindel分析软件得到最初的raw突变;然后经过重复区域、ssr、突变质量值、突变深度、突变率等严格过滤,得到高度可靠的突变,在dbSNP,OMIM,uniprot,ClinVar等数据库中进行检索和过滤,得到各个突变的疾病关联信息。
直接测序(Sanger法):验证首先对高通量测序所检测到的可疑致病基因及突变位点行生物信息学分析,确定突变的性质;其次对致病性突变进行遗传学分析,并与患儿的临床表型比较,选择临床表型较吻合的突变基因及其致病性突变位点进行验证。本文患儿发现的突变位点,首先利用Sanger直接测序的方法在患儿父母中进行突变位点验证。对新发现的错义突变进行在线PolyPhen和SFIT预测,并通过多物种蛋白序列比对分析新突变位点的保守性。
筛选流程见图2。
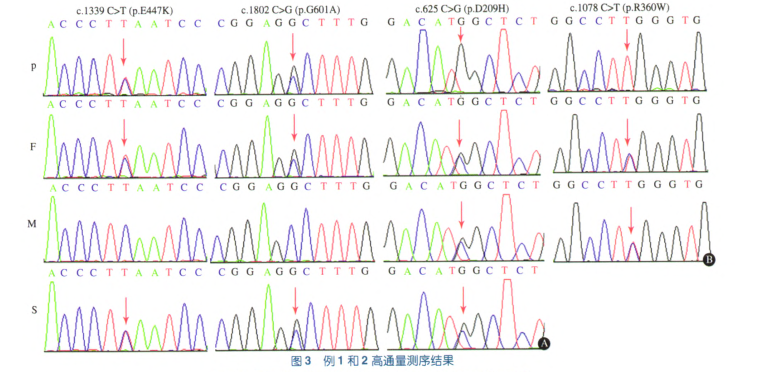
测序结果:4 000种单基因高通量测序和Sanger法验证发现,例l存在NPHSl基因已报道的P.E447K和P.G601A杂合突变,ADCK4基因纯合P.D209H错义突变,为新发现的突变。家系突变分析发现,NPHSl基因P.E447K和P.G601A杂合突变均来自父亲,其哥哥也有相同的基因型,其母亲不携带突变(图3A);患儿父母和哥哥分别携带杂合P.D209H错义突变(图3A)。例2检出COQ6基因的纯合P.R360W错义突变,为新发现的突变。家系突变分析显示,患儿父母分别携带杂合P.R360W错义突变(图3B)。2例患儿均确诊为线粒体相关肾病。
测序结果分析:ADCK4基因P.D209H突变和COQ6F M基因的p.R360W突变均为错义突变,经在线软件PolyPhen和SIFT预测为有害性突变。多物种蛋白序列比对发现,突变位点具有保守性。

治疗和随访
例1和2基因确诊后均予辅酶Q10 15—30 mg·kg-1·d“口服,例1随访6个月,尿蛋白+一2+,SCr 80~123 umol·L-1,BUN 6.0—7.0 mmol·L-1。例2随访6个月,尿蛋白3+一4+,SCr 19—23umol·L-1,BUN 3.6~4.0mmol·L-1,临床症状加重不明显。
线粒体病是最常见和最复杂的遗传性疾病,其遗传方式涉及到线粒体基因(mtDNA)的母系遗传和核基因(nDNA)的孟德尔遗传。各种遗传缺陷导致的线粒体结构和功能异常可引发多个系统能量代谢障碍,常累及脑、骨骼肌、眼、耳、消化、内分泌、心血管及血液系统等。近年来研究发现,多种线粒体基因或核基因遗传缺陷引起的线粒体功能障碍还可累及肾脏,称为线粒体相关。肾病,临床上对该病常认识不足。线粒体相关肾病可表现为肾小管功能障碍、间质性肾炎、囊性肾病变以及肾小球性病变。线粒体基因和线粒体蛋白编码的核基因突变均可引起肾小球病变,表现为蛋白尿或肾病综合征,既可伴有肾外症状,也可仅表现为肾小球受累,肾脏病理多为FSGS,对激素治疗及免疫抑制剂治疗无效,其中影响辅酶Q10合成基因突变的一部分患者对辅酶Q10治疗有效。目前线粒体相关。肾病在国内尚未见报道,国外报道也罕见。
线粒体相关肾病虽然罕见,但该病已经是儿童终末期肾病的重要病因之一。提高对该病的认识,早期发现和早期干预该病,有助于儿童慢性肾脏病的防止。
病例来源:爱爱医
版权声明:站所注明来源为"爱爱医"的文章,版权归作者与本站共同所有,非经授权不得转载。本站所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,如果您认为我们的转载侵犯了您的权益,请及时通过电话(400-626-9910)或邮箱(zlzs@120.net)通知我们,我们将第一时间处理,感谢。








全部评论
谢谢分享好病例 学生 学习了