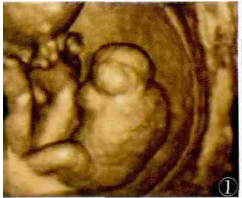
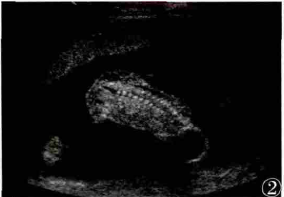
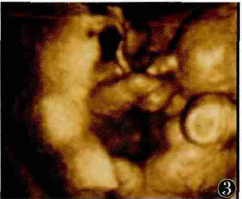
无心畸胎序列征1例
发布人:

4****5其他医务者
更新时间:2018-07-10 20:44
关注
【病案介绍】
主诉
【家族史】父母均体健,非近亲结婚,家族成员中无血液肿瘤疾病病史。
【讨论】
1985年,Stein等首先报道了间变性大细胞淋巴瘤(anaplastic large cell lymphoma,ALCL),ALCL是一种肿瘤细胞大而异型明显的外周T细胞淋巴瘤,免疫组化标记CD30强阳性,约有半数患者产生致癌性的异常间变性淋巴瘤激酶(anaplasticlymphoma kinase,ALK)融合蛋白。归纳国内外文献,ACLC特点如下:1.根据ALCL***的临床病理学类型,可将其分为两大类:原发系统型ALCL和原发皮肤型ALCL,前者为ALCL的主要类型,后者发病率极低,国内外均少见。2.根据细胞免疫表型,ALCL可来源于T细胞或裸细胞。3.根据细胞遗传学特征,ALCL存在t(2,5)染色体异常而表达ALK蛋白。原发系统型ALCL又依据ALK表达的情况,分为2个亚型:ALK阳性ALCL和ALK阴性ALCL。ALK阳性间变大细胞淋巴瘤是临床上少见的疾病,特别是原发性的病例更是罕见。4.临床表现:ALCL可累及淋巴结内外;多呈进展期疾病,可累及骨髓,患者可有高热等B症状,当发热消失后,患者往往无不适的临床表现,饮食可恢复正常。ALK阳性多见于儿童和年轻人,男、女比例为1.5~6.5:1,60%~70%就诊时为晚期,B症状及结外受累常见,常表现为浅表和腹腔淋巴结肿大,大肿块多见,占30%~54%,皮肤、骨和软组织是3个最常见的结外累及部位.治疗:原发系统性ALCL以化疗为主要治疗手段,儿童主要采用治疗淋巴母细胞白血病和(或)淋巴瘤的方案或Burkitt淋巴瘤的方案,并需预防中枢神经系统侵犯。6.预后:在ALCL的临床预后因素中,许多研究证实ALK阳性和阴性患者的预计5年OS率分别为85.9%和35.2%。前者的生存明显优于后者(P<o.01),进一步证实ALK表达可作为ALCL的***预后因素。ALK阳性者较ALK阴性者预后好,5年生存率可达80%,化疗敏感,ORR达92.3%,CR率为6l%。本例为儿童,不伴浅表淋巴结肿大,不累及骨髓,以泌尿系统表现就诊,后出现急性肾功能衰竭,继而发现腹腔淋巴结肿大及腰背部肿物。1个月前有高热和心包积液症状,发热消失后一般情况好,病程中LDH正常,肿瘤标志物阴性,血肌酐的水平也呈跳跃式变化,影像学检查提示引起梗阻的腹部结节大小和位置都有活动性变化。淋巴结,骨髓活检均未有特殊发现,为明确诊断行左侧腰背部肿块切除术,术后病理结果结合形态及免疫组化符合间变大细胞淋巴瘤,最终明确诊断。该患儿LDH正常,电解质正常,腹部肿块体积不大,肾功能衰竭不能用肿瘤溶解解释,与疾病本身的关系缺乏大样本资料。除2004年**成人和2008年美国成人各有1例相似的个案报道,间变大细胞淋巴瘤肾功能衰竭考虑输尿管梗阻压迫所致外,有无直接影响肾功能改变的机理尚待研究。根具有关资料分析,在引起输尿管梗阻的非结石性因素中,因恶性肿瘤引起的输尿管外压性梗阻占78%,最常见的恶性肿瘤依次为卵巢癌、淋巴瘤及宫颈癌。由于肿瘤直接压迫或侵犯输尿管,均可能引起输尿管机械性梗阻,导致输尿管扩张及肾积水,最终导致肾后性肾衰竭,严重威胁患者生命。对于这类患者,必须尽早明确诊断,针对不同病因及输尿管狭窄的部位、长度、狭窄的程度等情况制定合理有效的治疗方案。而MRU技术比较BUS超声及IVP检查具有扫描视野广、可清晰显示泌尿系统解剖结构、安全可靠、无需造影剂、无创伤性、无射线、准确率高、重复性好等优点,可以对小儿泌尿系梗阻作出明确诊断,为制订手术方案和估测预后提供可靠依据。由于ALCL临床结外表现的多样性及无特异性,诊断ALCL对病理学家和临床医师是一个挑战,ALCL仅依靠病理形态学诊断的准确性和可靠性仅为46%,如果形态学结合免疫表型(CD30阳性),诊断准确性提高到85%。因此ALK阳性ALCL诊断需根据组织病理学、细胞遗传学、免疫表型、分子生物学和临床表现,做出综合判断。关于ALCL发病机制的分子生物学研究,Morris等于1994年最先克隆了ALCL最常见的染色体异常,为t(2;5)(p23;q35),也即2号染色体上的ALK基因与5号染色体上的核磷小体(nucleophosmin,NPM)基因融合,易位产生NPM.ALK融合基因,从而产生具有持续性酪氨酸激酶活性的NPM.ALK融合蛋白,该融合蛋白可以激活多个与细胞生长有关的信号转导途径,从而导致肿瘤的发生。t(2;5)约占ALK阳性ALCL的75%,此外,还有15%-20%的患者出现其他的染色体异常,如t(1;2)(q25;p23),inV(2)(p23;q35),t(2;3)以及t(2;7)等,其预后与经典型t(2;5)患者的预后相似。对于复发难治的患者,采用单克隆靶向治疗、生物治疗及基因治疗等均是积极探索的方向。
来源:张文英, 陈椰. 以肾功能衰竭为首发症状的间变大细胞淋巴瘤1例[J]. 中国小儿血液与肿瘤杂志, 2017(5):269-271.
既往史
【家族史】父母均体健,非近亲结婚,家族成员中无血液肿瘤疾病病史。
【讨论】
1985年,Stein等首先报道了间变性大细胞淋巴瘤(anaplastic large cell lymphoma,ALCL),ALCL是一种肿瘤细胞大而异型明显的外周T细胞淋巴瘤,免疫组化标记CD30强阳性,约有半数患者产生致癌性的异常间变性淋巴瘤激酶(anaplasticlymphoma kinase,ALK)融合蛋白。归纳国内外文献,ACLC特点如下:1.根据ALCL***的临床病理学类型,可将其分为两大类:原发系统型ALCL和原发皮肤型ALCL,前者为ALCL的主要类型,后者发病率极低,国内外均少见。2.根据细胞免疫表型,ALCL可来源于T细胞或裸细胞。3.根据细胞遗传学特征,ALCL存在t(2,5)染色体异常而表达ALK蛋白。原发系统型ALCL又依据ALK表达的情况,分为2个亚型:ALK阳性ALCL和ALK阴性ALCL。ALK阳性间变大细胞淋巴瘤是临床上少见的疾病,特别是原发性的病例更是罕见。4.临床表现:ALCL可累及淋巴结内外;多呈进展期疾病,可累及骨髓,患者可有高热等B症状,当发热消失后,患者往往无不适的临床表现,饮食可恢复正常。ALK阳性多见于儿童和年轻人,男、女比例为1.5~6.5:1,60%~70%就诊时为晚期,B症状及结外受累常见,常表现为浅表和腹腔淋巴结肿大,大肿块多见,占30%~54%,皮肤、骨和软组织是3个最常见的结外累及部位.治疗:原发系统性ALCL以化疗为主要治疗手段,儿童主要采用治疗淋巴母细胞白血病和(或)淋巴瘤的方案或Burkitt淋巴瘤的方案,并需预防中枢神经系统侵犯。6.预后:在ALCL的临床预后因素中,许多研究证实ALK阳性和阴性患者的预计5年OS率分别为85.9%和35.2%。前者的生存明显优于后者(P<o.01),进一步证实ALK表达可作为ALCL的***预后因素。ALK阳性者较ALK阴性者预后好,5年生存率可达80%,化疗敏感,ORR达92.3%,CR率为6l%。本例为儿童,不伴浅表淋巴结肿大,不累及骨髓,以泌尿系统表现就诊,后出现急性肾功能衰竭,继而发现腹腔淋巴结肿大及腰背部肿物。1个月前有高热和心包积液症状,发热消失后一般情况好,病程中LDH正常,肿瘤标志物阴性,血肌酐的水平也呈跳跃式变化,影像学检查提示引起梗阻的腹部结节大小和位置都有活动性变化。淋巴结,骨髓活检均未有特殊发现,为明确诊断行左侧腰背部肿块切除术,术后病理结果结合形态及免疫组化符合间变大细胞淋巴瘤,最终明确诊断。该患儿LDH正常,电解质正常,腹部肿块体积不大,肾功能衰竭不能用肿瘤溶解解释,与疾病本身的关系缺乏大样本资料。除2004年**成人和2008年美国成人各有1例相似的个案报道,间变大细胞淋巴瘤肾功能衰竭考虑输尿管梗阻压迫所致外,有无直接影响肾功能改变的机理尚待研究。根具有关资料分析,在引起输尿管梗阻的非结石性因素中,因恶性肿瘤引起的输尿管外压性梗阻占78%,最常见的恶性肿瘤依次为卵巢癌、淋巴瘤及宫颈癌。由于肿瘤直接压迫或侵犯输尿管,均可能引起输尿管机械性梗阻,导致输尿管扩张及肾积水,最终导致肾后性肾衰竭,严重威胁患者生命。对于这类患者,必须尽早明确诊断,针对不同病因及输尿管狭窄的部位、长度、狭窄的程度等情况制定合理有效的治疗方案。而MRU技术比较BUS超声及IVP检查具有扫描视野广、可清晰显示泌尿系统解剖结构、安全可靠、无需造影剂、无创伤性、无射线、准确率高、重复性好等优点,可以对小儿泌尿系梗阻作出明确诊断,为制订手术方案和估测预后提供可靠依据。由于ALCL临床结外表现的多样性及无特异性,诊断ALCL对病理学家和临床医师是一个挑战,ALCL仅依靠病理形态学诊断的准确性和可靠性仅为46%,如果形态学结合免疫表型(CD30阳性),诊断准确性提高到85%。因此ALK阳性ALCL诊断需根据组织病理学、细胞遗传学、免疫表型、分子生物学和临床表现,做出综合判断。关于ALCL发病机制的分子生物学研究,Morris等于1994年最先克隆了ALCL最常见的染色体异常,为t(2;5)(p23;q35),也即2号染色体上的ALK基因与5号染色体上的核磷小体(nucleophosmin,NPM)基因融合,易位产生NPM.ALK融合基因,从而产生具有持续性酪氨酸激酶活性的NPM.ALK融合蛋白,该融合蛋白可以激活多个与细胞生长有关的信号转导途径,从而导致肿瘤的发生。t(2;5)约占ALK阳性ALCL的75%,此外,还有15%-20%的患者出现其他的染色体异常,如t(1;2)(q25;p23),inV(2)(p23;q35),t(2;3)以及t(2;7)等,其预后与经典型t(2;5)患者的预后相似。对于复发难治的患者,采用单克隆靶向治疗、生物治疗及基因治疗等均是积极探索的方向。
来源:张文英, 陈椰. 以肾功能衰竭为首发症状的间变大细胞淋巴瘤1例[J]. 中国小儿血液与肿瘤杂志, 2017(5):269-271.
辅助检查
【家族史】父母均体健,非近亲结婚,家族成员中无血液肿瘤疾病病史。
【讨论】
1985年,Stein等首先报道了间变性大细胞淋巴瘤(anaplastic large cell lymphoma,ALCL),ALCL是一种肿瘤细胞大而异型明显的外周T细胞淋巴瘤,免疫组化标记CD30强阳性,约有半数患者产生致癌性的异常间变性淋巴瘤激酶(anaplasticlymphoma kinase,ALK)融合蛋白。归纳国内外文献,ACLC特点如下:1.根据ALCL***的临床病理学类型,可将其分为两大类:原发系统型ALCL和原发皮肤型ALCL,前者为ALCL的主要类型,后者发病率极低,国内外均少见。2.根据细胞免疫表型,ALCL可来源于T细胞或裸细胞。3.根据细胞遗传学特征,ALCL存在t(2,5)染色体异常而表达ALK蛋白。原发系统型ALCL又依据ALK表达的情况,分为2个亚型:ALK阳性ALCL和ALK阴性ALCL。ALK阳性间变大细胞淋巴瘤是临床上少见的疾病,特别是原发性的病例更是罕见。4.临床表现:ALCL可累及淋巴结内外;多呈进展期疾病,可累及骨髓,患者可有高热等B症状,当发热消失后,患者往往无不适的临床表现,饮食可恢复正常。ALK阳性多见于儿童和年轻人,男、女比例为1.5~6.5:1,60%~70%就诊时为晚期,B症状及结外受累常见,常表现为浅表和腹腔淋巴结肿大,大肿块多见,占30%~54%,皮肤、骨和软组织是3个最常见的结外累及部位.治疗:原发系统性ALCL以化疗为主要治疗手段,儿童主要采用治疗淋巴母细胞白血病和(或)淋巴瘤的方案或Burkitt淋巴瘤的方案,并需预防中枢神经系统侵犯。6.预后:在ALCL的临床预后因素中,许多研究证实ALK阳性和阴性患者的预计5年OS率分别为85.9%和35.2%。前者的生存明显优于后者(P<o.01),进一步证实ALK表达可作为ALCL的***预后因素。ALK阳性者较ALK阴性者预后好,5年生存率可达80%,化疗敏感,ORR达92.3%,CR率为6l%。本例为儿童,不伴浅表淋巴结肿大,不累及骨髓,以泌尿系统表现就诊,后出现急性肾功能衰竭,继而发现腹腔淋巴结肿大及腰背部肿物。1个月前有高热和心包积液症状,发热消失后一般情况好,病程中LDH正常,肿瘤标志物阴性,血肌酐的水平也呈跳跃式变化,影像学检查提示引起梗阻的腹部结节大小和位置都有活动性变化。淋巴结,骨髓活检均未有特殊发现,为明确诊断行左侧腰背部肿块切除术,术后病理结果结合形态及免疫组化符合间变大细胞淋巴瘤,最终明确诊断。该患儿LDH正常,电解质正常,腹部肿块体积不大,肾功能衰竭不能用肿瘤溶解解释,与疾病本身的关系缺乏大样本资料。除2004年**成人和2008年美国成人各有1例相似的个案报道,间变大细胞淋巴瘤肾功能衰竭考虑输尿管梗阻压迫所致外,有无直接影响肾功能改变的机理尚待研究。根具有关资料分析,在引起输尿管梗阻的非结石性因素中,因恶性肿瘤引起的输尿管外压性梗阻占78%,最常见的恶性肿瘤依次为卵巢癌、淋巴瘤及宫颈癌。由于肿瘤直接压迫或侵犯输尿管,均可能引起输尿管机械性梗阻,导致输尿管扩张及肾积水,最终导致肾后性肾衰竭,严重威胁患者生命。对于这类患者,必须尽早明确诊断,针对不同病因及输尿管狭窄的部位、长度、狭窄的程度等情况制定合理有效的治疗方案。而MRU技术比较BUS超声及IVP检查具有扫描视野广、可清晰显示泌尿系统解剖结构、安全可靠、无需造影剂、无创伤性、无射线、准确率高、重复性好等优点,可以对小儿泌尿系梗阻作出明确诊断,为制订手术方案和估测预后提供可靠依据。由于ALCL临床结外表现的多样性及无特异性,诊断ALCL对病理学家和临床医师是一个挑战,ALCL仅依靠病理形态学诊断的准确性和可靠性仅为46%,如果形态学结合免疫表型(CD30阳性),诊断准确性提高到85%。因此ALK阳性ALCL诊断需根据组织病理学、细胞遗传学、免疫表型、分子生物学和临床表现,做出综合判断。关于ALCL发病机制的分子生物学研究,Morris等于1994年最先克隆了ALCL最常见的染色体异常,为t(2;5)(p23;q35),也即2号染色体上的ALK基因与5号染色体上的核磷小体(nucleophosmin,NPM)基因融合,易位产生NPM.ALK融合基因,从而产生具有持续性酪氨酸激酶活性的NPM.ALK融合蛋白,该融合蛋白可以激活多个与细胞生长有关的信号转导途径,从而导致肿瘤的发生。t(2;5)约占ALK阳性ALCL的75%,此外,还有15%-20%的患者出现其他的染色体异常,如t(1;2)(q25;p23),inV(2)(p23;q35),t(2;3)以及t(2;7)等,其预后与经典型t(2;5)患者的预后相似。对于复发难治的患者,采用单克隆靶向治疗、生物治疗及基因治疗等均是积极探索的方向。
来源:张文英, 陈椰. 以肾功能衰竭为首发症状的间变大细胞淋巴瘤1例[J]. 中国小儿血液与肿瘤杂志, 2017(5):269-271.
【诊治过程】
初步诊断
【家族史】父母均体健,非近亲结婚,家族成员中无血液肿瘤疾病病史。
【讨论】
1985年,Stein等首先报道了间变性大细胞淋巴瘤(anaplastic large cell lymphoma,ALCL),ALCL是一种肿瘤细胞大而异型明显的外周T细胞淋巴瘤,免疫组化标记CD30强阳性,约有半数患者产生致癌性的异常间变性淋巴瘤激酶(anaplasticlymphoma kinase,ALK)融合蛋白。归纳国内外文献,ACLC特点如下:1.根据ALCL***的临床病理学类型,可将其分为两大类:原发系统型ALCL和原发皮肤型ALCL,前者为ALCL的主要类型,后者发病率极低,国内外均少见。2.根据细胞免疫表型,ALCL可来源于T细胞或裸细胞。3.根据细胞遗传学特征,ALCL存在t(2,5)染色体异常而表达ALK蛋白。原发系统型ALCL又依据ALK表达的情况,分为2个亚型:ALK阳性ALCL和ALK阴性ALCL。ALK阳性间变大细胞淋巴瘤是临床上少见的疾病,特别是原发性的病例更是罕见。4.临床表现:ALCL可累及淋巴结内外;多呈进展期疾病,可累及骨髓,患者可有高热等B症状,当发热消失后,患者往往无不适的临床表现,饮食可恢复正常。ALK阳性多见于儿童和年轻人,男、女比例为1.5~6.5:1,60%~70%就诊时为晚期,B症状及结外受累常见,常表现为浅表和腹腔淋巴结肿大,大肿块多见,占30%~54%,皮肤、骨和软组织是3个最常见的结外累及部位.治疗:原发系统性ALCL以化疗为主要治疗手段,儿童主要采用治疗淋巴母细胞白血病和(或)淋巴瘤的方案或Burkitt淋巴瘤的方案,并需预防中枢神经系统侵犯。6.预后:在ALCL的临床预后因素中,许多研究证实ALK阳性和阴性患者的预计5年OS率分别为85.9%和35.2%。前者的生存明显优于后者(P<o.01),进一步证实ALK表达可作为ALCL的***预后因素。ALK阳性者较ALK阴性者预后好,5年生存率可达80%,化疗敏感,ORR达92.3%,CR率为6l%。本例为儿童,不伴浅表淋巴结肿大,不累及骨髓,以泌尿系统表现就诊,后出现急性肾功能衰竭,继而发现腹腔淋巴结肿大及腰背部肿物。1个月前有高热和心包积液症状,发热消失后一般情况好,病程中LDH正常,肿瘤标志物阴性,血肌酐的水平也呈跳跃式变化,影像学检查提示引起梗阻的腹部结节大小和位置都有活动性变化。淋巴结,骨髓活检均未有特殊发现,为明确诊断行左侧腰背部肿块切除术,术后病理结果结合形态及免疫组化符合间变大细胞淋巴瘤,最终明确诊断。该患儿LDH正常,电解质正常,腹部肿块体积不大,肾功能衰竭不能用肿瘤溶解解释,与疾病本身的关系缺乏大样本资料。除2004年**成人和2008年美国成人各有1例相似的个案报道,间变大细胞淋巴瘤肾功能衰竭考虑输尿管梗阻压迫所致外,有无直接影响肾功能改变的机理尚待研究。根具有关资料分析,在引起输尿管梗阻的非结石性因素中,因恶性肿瘤引起的输尿管外压性梗阻占78%,最常见的恶性肿瘤依次为卵巢癌、淋巴瘤及宫颈癌。由于肿瘤直接压迫或侵犯输尿管,均可能引起输尿管机械性梗阻,导致输尿管扩张及肾积水,最终导致肾后性肾衰竭,严重威胁患者生命。对于这类患者,必须尽早明确诊断,针对不同病因及输尿管狭窄的部位、长度、狭窄的程度等情况制定合理有效的治疗方案。而MRU技术比较BUS超声及IVP检查具有扫描视野广、可清晰显示泌尿系统解剖结构、安全可靠、无需造影剂、无创伤性、无射线、准确率高、重复性好等优点,可以对小儿泌尿系梗阻作出明确诊断,为制订手术方案和估测预后提供可靠依据。由于ALCL临床结外表现的多样性及无特异性,诊断ALCL对病理学家和临床医师是一个挑战,ALCL仅依靠病理形态学诊断的准确性和可靠性仅为46%,如果形态学结合免疫表型(CD30阳性),诊断准确性提高到85%。因此ALK阳性ALCL诊断需根据组织病理学、细胞遗传学、免疫表型、分子生物学和临床表现,做出综合判断。关于ALCL发病机制的分子生物学研究,Morris等于1994年最先克隆了ALCL最常见的染色体异常,为t(2;5)(p23;q35),也即2号染色体上的ALK基因与5号染色体上的核磷小体(nucleophosmin,NPM)基因融合,易位产生NPM.ALK融合基因,从而产生具有持续性酪氨酸激酶活性的NPM.ALK融合蛋白,该融合蛋白可以激活多个与细胞生长有关的信号转导途径,从而导致肿瘤的发生。t(2;5)约占ALK阳性ALCL的75%,此外,还有15%-20%的患者出现其他的染色体异常,如t(1;2)(q25;p23),inV(2)(p23;q35),t(2;3)以及t(2;7)等,其预后与经典型t(2;5)患者的预后相似。对于复发难治的患者,采用单克隆靶向治疗、生物治疗及基因治疗等均是积极探索的方向。
来源:张文英, 陈椰. 以肾功能衰竭为首发症状的间变大细胞淋巴瘤1例[J]. 中国小儿血液与肿瘤杂志, 2017(5):269-271.
诊治经过
【家族史】父母均体健,非近亲结婚,家族成员中无血液肿瘤疾病病史。
【讨论】
1985年,Stein等首先报道了间变性大细胞淋巴瘤(anaplastic large cell lymphoma,ALCL),ALCL是一种肿瘤细胞大而异型明显的外周T细胞淋巴瘤,免疫组化标记CD30强阳性,约有半数患者产生致癌性的异常间变性淋巴瘤激酶(anaplasticlymphoma kinase,ALK)融合蛋白。归纳国内外文献,ACLC特点如下:1.根据ALCL***的临床病理学类型,可将其分为两大类:原发系统型ALCL和原发皮肤型ALCL,前者为ALCL的主要类型,后者发病率极低,国内外均少见。2.根据细胞免疫表型,ALCL可来源于T细胞或裸细胞。3.根据细胞遗传学特征,ALCL存在t(2,5)染色体异常而表达ALK蛋白。原发系统型ALCL又依据ALK表达的情况,分为2个亚型:ALK阳性ALCL和ALK阴性ALCL。ALK阳性间变大细胞淋巴瘤是临床上少见的疾病,特别是原发性的病例更是罕见。4.临床表现:ALCL可累及淋巴结内外;多呈进展期疾病,可累及骨髓,患者可有高热等B症状,当发热消失后,患者往往无不适的临床表现,饮食可恢复正常。ALK阳性多见于儿童和年轻人,男、女比例为1.5~6.5:1,60%~70%就诊时为晚期,B症状及结外受累常见,常表现为浅表和腹腔淋巴结肿大,大肿块多见,占30%~54%,皮肤、骨和软组织是3个最常见的结外累及部位.治疗:原发系统性ALCL以化疗为主要治疗手段,儿童主要采用治疗淋巴母细胞白血病和(或)淋巴瘤的方案或Burkitt淋巴瘤的方案,并需预防中枢神经系统侵犯。6.预后:在ALCL的临床预后因素中,许多研究证实ALK阳性和阴性患者的预计5年OS率分别为85.9%和35.2%。前者的生存明显优于后者(P<o.01),进一步证实ALK表达可作为ALCL的***预后因素。ALK阳性者较ALK阴性者预后好,5年生存率可达80%,化疗敏感,ORR达92.3%,CR率为6l%。本例为儿童,不伴浅表淋巴结肿大,不累及骨髓,以泌尿系统表现就诊,后出现急性肾功能衰竭,继而发现腹腔淋巴结肿大及腰背部肿物。1个月前有高热和心包积液症状,发热消失后一般情况好,病程中LDH正常,肿瘤标志物阴性,血肌酐的水平也呈跳跃式变化,影像学检查提示引起梗阻的腹部结节大小和位置都有活动性变化。淋巴结,骨髓活检均未有特殊发现,为明确诊断行左侧腰背部肿块切除术,术后病理结果结合形态及免疫组化符合间变大细胞淋巴瘤,最终明确诊断。该患儿LDH正常,电解质正常,腹部肿块体积不大,肾功能衰竭不能用肿瘤溶解解释,与疾病本身的关系缺乏大样本资料。除2004年**成人和2008年美国成人各有1例相似的个案报道,间变大细胞淋巴瘤肾功能衰竭考虑输尿管梗阻压迫所致外,有无直接影响肾功能改变的机理尚待研究。根具有关资料分析,在引起输尿管梗阻的非结石性因素中,因恶性肿瘤引起的输尿管外压性梗阻占78%,最常见的恶性肿瘤依次为卵巢癌、淋巴瘤及宫颈癌。由于肿瘤直接压迫或侵犯输尿管,均可能引起输尿管机械性梗阻,导致输尿管扩张及肾积水,最终导致肾后性肾衰竭,严重威胁患者生命。对于这类患者,必须尽早明确诊断,针对不同病因及输尿管狭窄的部位、长度、狭窄的程度等情况制定合理有效的治疗方案。而MRU技术比较BUS超声及IVP检查具有扫描视野广、可清晰显示泌尿系统解剖结构、安全可靠、无需造影剂、无创伤性、无射线、准确率高、重复性好等优点,可以对小儿泌尿系梗阻作出明确诊断,为制订手术方案和估测预后提供可靠依据。由于ALCL临床结外表现的多样性及无特异性,诊断ALCL对病理学家和临床医师是一个挑战,ALCL仅依靠病理形态学诊断的准确性和可靠性仅为46%,如果形态学结合免疫表型(CD30阳性),诊断准确性提高到85%。因此ALK阳性ALCL诊断需根据组织病理学、细胞遗传学、免疫表型、分子生物学和临床表现,做出综合判断。关于ALCL发病机制的分子生物学研究,Morris等于1994年最先克隆了ALCL最常见的染色体异常,为t(2;5)(p23;q35),也即2号染色体上的ALK基因与5号染色体上的核磷小体(nucleophosmin,NPM)基因融合,易位产生NPM.ALK融合基因,从而产生具有持续性酪氨酸激酶活性的NPM.ALK融合蛋白,该融合蛋白可以激活多个与细胞生长有关的信号转导途径,从而导致肿瘤的发生。t(2;5)约占ALK阳性ALCL的75%,此外,还有15%-20%的患者出现其他的染色体异常,如t(1;2)(q25;p23),inV(2)(p23;q35),t(2;3)以及t(2;7)等,其预后与经典型t(2;5)患者的预后相似。对于复发难治的患者,采用单克隆靶向治疗、生物治疗及基因治疗等均是积极探索的方向。
来源:张文英, 陈椰. 以肾功能衰竭为首发症状的间变大细胞淋巴瘤1例[J]. 中国小儿血液与肿瘤杂志, 2017(5):269-271.
【其他】
【家族史】父母均体健,非近亲结婚,家族成员中无血液肿瘤疾病病史。
【讨论】
1985年,Stein等首先报道了间变性大细胞淋巴瘤(anaplastic large cell lymphoma,ALCL),ALCL是一种肿瘤细胞大而异型明显的外周T细胞淋巴瘤,免疫组化标记CD30强阳性,约有半数患者产生致癌性的异常间变性淋巴瘤激酶(anaplasticlymphoma kinase,ALK)融合蛋白。归纳国内外文献,ACLC特点如下:1.根据ALCL***的临床病理学类型,可将其分为两大类:原发系统型ALCL和原发皮肤型ALCL,前者为ALCL的主要类型,后者发病率极低,国内外均少见。2.根据细胞免疫表型,ALCL可来源于T细胞或裸细胞。3.根据细胞遗传学特征,ALCL存在t(2,5)染色体异常而表达ALK蛋白。原发系统型ALCL又依据ALK表达的情况,分为2个亚型:ALK阳性ALCL和ALK阴性ALCL。ALK阳性间变大细胞淋巴瘤是临床上少见的疾病,特别是原发性的病例更是罕见。4.临床表现:ALCL可累及淋巴结内外;多呈进展期疾病,可累及骨髓,患者可有高热等B症状,当发热消失后,患者往往无不适的临床表现,饮食可恢复正常。ALK阳性多见于儿童和年轻人,男、女比例为1.5~6.5:1,60%~70%就诊时为晚期,B症状及结外受累常见,常表现为浅表和腹腔淋巴结肿大,大肿块多见,占30%~54%,皮肤、骨和软组织是3个最常见的结外累及部位.治疗:原发系统性ALCL以化疗为主要治疗手段,儿童主要采用治疗淋巴母细胞白血病和(或)淋巴瘤的方案或Burkitt淋巴瘤的方案,并需预防中枢神经系统侵犯。6.预后:在ALCL的临床预后因素中,许多研究证实ALK阳性和阴性患者的预计5年OS率分别为85.9%和35.2%。前者的生存明显优于后者(P<o.01),进一步证实ALK表达可作为ALCL的***预后因素。ALK阳性者较ALK阴性者预后好,5年生存率可达80%,化疗敏感,ORR达92.3%,CR率为6l%。本例为儿童,不伴浅表淋巴结肿大,不累及骨髓,以泌尿系统表现就诊,后出现急性肾功能衰竭,继而发现腹腔淋巴结肿大及腰背部肿物。1个月前有高热和心包积液症状,发热消失后一般情况好,病程中LDH正常,肿瘤标志物阴性,血肌酐的水平也呈跳跃式变化,影像学检查提示引起梗阻的腹部结节大小和位置都有活动性变化。淋巴结,骨髓活检均未有特殊发现,为明确诊断行左侧腰背部肿块切除术,术后病理结果结合形态及免疫组化符合间变大细胞淋巴瘤,最终明确诊断。该患儿LDH正常,电解质正常,腹部肿块体积不大,肾功能衰竭不能用肿瘤溶解解释,与疾病本身的关系缺乏大样本资料。除2004年**成人和2008年美国成人各有1例相似的个案报道,间变大细胞淋巴瘤肾功能衰竭考虑输尿管梗阻压迫所致外,有无直接影响肾功能改变的机理尚待研究。根具有关资料分析,在引起输尿管梗阻的非结石性因素中,因恶性肿瘤引起的输尿管外压性梗阻占78%,最常见的恶性肿瘤依次为卵巢癌、淋巴瘤及宫颈癌。由于肿瘤直接压迫或侵犯输尿管,均可能引起输尿管机械性梗阻,导致输尿管扩张及肾积水,最终导致肾后性肾衰竭,严重威胁患者生命。对于这类患者,必须尽早明确诊断,针对不同病因及输尿管狭窄的部位、长度、狭窄的程度等情况制定合理有效的治疗方案。而MRU技术比较BUS超声及IVP检查具有扫描视野广、可清晰显示泌尿系统解剖结构、安全可靠、无需造影剂、无创伤性、无射线、准确率高、重复性好等优点,可以对小儿泌尿系梗阻作出明确诊断,为制订手术方案和估测预后提供可靠依据。由于ALCL临床结外表现的多样性及无特异性,诊断ALCL对病理学家和临床医师是一个挑战,ALCL仅依靠病理形态学诊断的准确性和可靠性仅为46%,如果形态学结合免疫表型(CD30阳性),诊断准确性提高到85%。因此ALK阳性ALCL诊断需根据组织病理学、细胞遗传学、免疫表型、分子生物学和临床表现,做出综合判断。关于ALCL发病机制的分子生物学研究,Morris等于1994年最先克隆了ALCL最常见的染色体异常,为t(2;5)(p23;q35),也即2号染色体上的ALK基因与5号染色体上的核磷小体(nucleophosmin,NPM)基因融合,易位产生NPM.ALK融合基因,从而产生具有持续性酪氨酸激酶活性的NPM.ALK融合蛋白,该融合蛋白可以激活多个与细胞生长有关的信号转导途径,从而导致肿瘤的发生。t(2;5)约占ALK阳性ALCL的75%,此外,还有15%-20%的患者出现其他的染色体异常,如t(1;2)(q25;p23),inV(2)(p23;q35),t(2;3)以及t(2;7)等,其预后与经典型t(2;5)患者的预后相似。对于复发难治的患者,采用单克隆靶向治疗、生物治疗及基因治疗等均是积极探索的方向。
来源:张文英, 陈椰. 以肾功能衰竭为首发症状的间变大细胞淋巴瘤1例[J]. 中国小儿血液与肿瘤杂志, 2017(5):269-271.
病例来源:爱爱医
版权声明:站所注明来源为"爱爱医"的文章,版权归作者与本站共同所有,非经授权不得转载。本站所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,如果您认为我们的转载侵犯了您的权益,请及时通过电话(400-626-9910)或邮箱(zlzs@120.net)通知我们,我们将第一时间处理,感谢。







全部评论