【病案介绍】
主诉
【讨论】
3.1发病概况及病因目前尚未有关于CD发病率的文献报道。文献报道国外患者平均年龄为35.6岁,国内为33.1岁。其发病率以女性多见,男∶女= 1∶4,但亦有文献报道发病率无性别差异。CD在临床上可以分为局灶型(Localized or unicentric CD,LCD orUCD)和多中心型(multicentric CD,MCD)。局灶型多发于青年,中位年龄约20岁,其中90%的局灶型病例为透明血管型,10%为浆细胞型。多中心型较局灶型少见,中位年龄57岁,男女发病率为1∶2。该病可发生在淋巴结存在的任何部位,国内报道以胸部的纵隔最多见(60%~70%),其次为颈部(10%~14%),腹部(5%~10%),腋部(2%~4%)等。国外报道局灶型好发部位为纵膈(30%),颈部(23%),腹部(20%),腹膜后腔(17%)及腋窝(5%),腹股沟(3%)和盆腔(2%)。既往对CD的病因研究认为,γ疱疹病毒HHV-8感染,血清白细胞介素6水平上升,VEGF引导的血管增生及滤泡树突状细胞不典型增生与CD的发生发展存在一定的相关性。3.2临床表现UCD患者常常没有自觉症状,于影像学检查时意外发现无痛性肿块。少部分患者可出现咳嗽,呼吸困难,或者其他全身症状。在大部分的UCD患者中表现为非进展性的临床表现。与此不同的是,MCD伴随全身炎症反应的系统表现:发热,盗汗,体重减轻,疲乏。MCD 患者常常有外周的淋巴结病,影响同一区域的多个淋巴结,实验室检测结果异常,比如:贫血、低蛋白血症、高丙种球蛋白血症和ESR、IL-6、IL-10水平升高。当以上的临床表现及实验室检查异常同时出现时,常常提示MCD的可能。影像学检查多表现为占位性病变或淋巴结肿大,无明显特征,发生于纵膈淋巴结的Castleman病在胸片上多表现为高密度团块影,边缘规整或有分叶,可单发或者多发,常见纵膈增宽的间接表现,累及肺实质。超声检查多表现为回声均匀的低回声团块影,包膜清晰完整;较大的病灶(大于5 cm)可表现为混杂不均匀的低回声,包膜不均匀。CT检查多表现为软组织肿块影,在动态扫描的变化过程类似于动脉。MRI 检查多表现为T1加权像上低信号和T2加权像上高信号影。MCD在影像学检查中还可见肝脾肿大及腹腔或胸腔积液表现。本病例表现为右侧颈部Ⅱ区占位性病变,内部结构较为一致,边界较清,与周边无明显粘连,呈良性肿物的影像学表现,无特征性改变。3.3诊断与鉴别诊断由于本病发病部位不同,临床表现多样化,且临床症状无特异性,故术前诊断较困难,术前CT检查对确立本病有一定帮助,平扫显示病灶为密度均匀,边缘光整清楚或不光整软组织肿块,CT增强扫描,透明血管型UCD病灶强化程度稍稍低于或与邻近大血管同步明显强化,肿块明显强化是由于肿块内有较多血管和丰富毛细血管所致,而浆细胞UCD因血管成分较少呈不均匀轻中度强化,缺少特征表现;MCD其CT表现无明显肿块,往往表现一个或多个区域大小相似的淋巴结,增强呈轻中度强化。后两种情况,影像学表现不典型,术前诊断较困难。本病的早期确诊最终有赖于组织病理学检查。对于CD,不管临床或病理分类,都有组织学的共同特点:①具有完整的淋巴结基本结构;②淋巴滤泡和血管增生,在透明血管型滤泡、血管发生玻璃样变,滤泡生发中心萎缩;浆细胞型表现滤泡间质中有较多的浆细胞,滤泡生发中心增生。Frizzera提出CD的诊断标准:①UCD的诊断标准:单一部位淋巴结肿大,组织病理学上具有特征性增生,除外可能的其他原发病,多无全身症状及贫血、免疫球蛋白升高等(浆细胞型除外),肿物切除后长期存活;②MCD诊断标准:具有特征性组织病理学改变,显著淋巴结肿大累及多处外周淋巴结,多系统受累表现,排除其他病因。在鉴别诊断上,应与恶性淋巴瘤、淋巴结核、非特异性淋巴结炎等鉴别。与恶性淋巴瘤的鉴别包括以下几点:恶性淋巴瘤时淋巴结结构几乎完全被破坏,少有血管增生,具有单克隆的淋巴细胞或R-S细胞增生。对于常见的以淋巴结肿大为表现的疾病,如非特异性淋巴结炎及淋巴结核,两者鉴别要点在于:①体格检查上,前者多有按压痛而后者无明显按压痛;②血液生化检查可查及前者血液中中性粒细胞水平上升;③PPD试验鉴别。3.4治疗及预后基于既往的研究,CD应采取切除异常淋巴结的外科治疗方式。对于UCD,切除病变淋巴结是最为有效的方式。国外学者的临床追踪表明,UCD经过手术治疗后,基本没有复发倾向。值得一提的是,有全身症状的患者在切除病变淋巴结后,症状消失。但也有个别报道指出,外科手术治疗后9年,患者出现UCD复发。近年来的一个以278份病例报告为基础的META分析结果,得出:UCD外科手术治疗患者的死亡率为3.8%,而其他治疗方式为17.6%。因此,外科手段虽然十分有效,但也有失败复发的风险。无法完全切除的病变,剩余的部分将会进展,更多的此类病例则是保持或者无明显症状表现。利妥昔单抗是治疗MCD的药物,也尝试用于不能清除的UCD患者身上,并获得良好效果。至于MCD,因为受到检查方式的限制,外科切除不是主要的治疗方式。免疫治疗及化学疗法是MCD的基本治疗策略。MCD的预后评估也通常比UCD更为保守。因为现在还没有完整的MCD治疗方式的随机试验研究,治疗方式的选择评估受限于现有的病例报告。利妥昔单抗单独应用或者联合细胞抑制剂应用已经在许多患者身上取得了效果。利妥昔单抗的临床反应包括血清IL-6水平的下降以及其他HIV阳性患者病例中,随着HHV-8病毒水平下降而下降的细胞因子。在更严重的疾病情况下,出现了器官衰竭和体力下降的征象时,利妥昔单抗联合依托泊苷等化疗药物,可以提高5年生存率到90%。然而,化疗药物必须在组合方案中应用:单抗疗法联合长春花碱或者依托泊苷可以抑制MCD,但是一旦停用后,症状将会在数周内复发。最新的化疗方案,比如CHOP(cyclophosphamide,doxo-rubicin,vincristine,and prednisone),即环磷酰胺+多柔比星+长春新碱+**或者CVAD(cyclo-phosvnamide,pirarubicin,vincristine and dexameth-asone),即环磷酰胺+ 吡柔比星+长春新碱+**,在50%以上的患者中展示出持久的疗效。3.5相关疾病病程虽然CD是以淋巴结病变为主的疾病,但是无论UCD和MCD都有可能伴发着其他疾病发生。在法国的一系列病例中,8名UCD患者伴发恶性淋巴瘤,其中6位为B细胞非霍奇金淋巴瘤,2例为霍奇金淋巴瘤。8例患者中3例同时出现UCD和NHL的发病,即便前者与后者之间的平均诊断间隔为46个月,两者间的病理生理学关系尚未明了。除此之外,接近15% UCD患者发展出副肿瘤性天疱疮。MCD患者有相似的伴发疾病,而且比UCD患者有更多的出现多脏器功能紊乱的概率。例如,MCD患者有更高的恶性肿瘤的发病风险,比如卡波氏肉瘤和血液系统恶性肿瘤。13%MCD患者同时患有卡波氏肉瘤。NHL与MCD有显著的关系:15%到20%的患者伴发或者发展出NHL,预后较差,死亡率高达85%。HHV-联合HIV感染,被认为是MCD发展为NHL重要的风险因素。弥漫性大B细胞性淋巴瘤是HHV-阳性MCD患者最常见的淋巴瘤,这也是2008年WHO淋巴组织来源的恶性肿瘤分类中独特的一种。虽然潜在的机制尚未能确定,但霍奇金病可能与MCD 有关。MCD也伴随着POEMS 综合征,即多发性神经病变(polyneuropathy,P)、脏器肿大(organomegaly,O)、内分泌病变(endocrinopathy,E),单克隆γ球蛋白病(monoclonal gammopathy,M,也叫M-蛋白)和皮肤改变(skinchanges,S)。
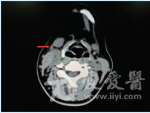
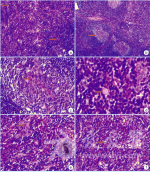
来源:热娜古孜·阿不都米吉提,李世豪, 黄子贤,等. 颈部Castleman病临床报道暨文献回顾[J]. 口腔疾病防止, 2017, 25(2):110-114.
现病史
【讨论】
3.1发病概况及病因目前尚未有关于CD发病率的文献报道。文献报道国外患者平均年龄为35.6岁,国内为33.1岁。其发病率以女性多见,男∶女= 1∶4,但亦有文献报道发病率无性别差异。CD在临床上可以分为局灶型(Localized or unicentric CD,LCD orUCD)和多中心型(multicentric CD,MCD)。局灶型多发于青年,中位年龄约20岁,其中90%的局灶型病例为透明血管型,10%为浆细胞型。多中心型较局灶型少见,中位年龄57岁,男女发病率为1∶2。该病可发生在淋巴结存在的任何部位,国内报道以胸部的纵隔最多见(60%~70%),其次为颈部(10%~14%),腹部(5%~10%),腋部(2%~4%)等。国外报道局灶型好发部位为纵膈(30%),颈部(23%),腹部(20%),腹膜后腔(17%)及腋窝(5%),腹股沟(3%)和盆腔(2%)。既往对CD的病因研究认为,γ疱疹病毒HHV-8感染,血清白细胞介素6水平上升,VEGF引导的血管增生及滤泡树突状细胞不典型增生与CD的发生发展存在一定的相关性。3.2临床表现UCD患者常常没有自觉症状,于影像学检查时意外发现无痛性肿块。少部分患者可出现咳嗽,呼吸困难,或者其他全身症状。在大部分的UCD患者中表现为非进展性的临床表现。与此不同的是,MCD伴随全身炎症反应的系统表现:发热,盗汗,体重减轻,疲乏。MCD 患者常常有外周的淋巴结病,影响同一区域的多个淋巴结,实验室检测结果异常,比如:贫血、低蛋白血症、高丙种球蛋白血症和ESR、IL-6、IL-10水平升高。当以上的临床表现及实验室检查异常同时出现时,常常提示MCD的可能。影像学检查多表现为占位性病变或淋巴结肿大,无明显特征,发生于纵膈淋巴结的Castleman病在胸片上多表现为高密度团块影,边缘规整或有分叶,可单发或者多发,常见纵膈增宽的间接表现,累及肺实质。超声检查多表现为回声均匀的低回声团块影,包膜清晰完整;较大的病灶(大于5 cm)可表现为混杂不均匀的低回声,包膜不均匀。CT检查多表现为软组织肿块影,在动态扫描的变化过程类似于动脉。MRI 检查多表现为T1加权像上低信号和T2加权像上高信号影。MCD在影像学检查中还可见肝脾肿大及腹腔或胸腔积液表现。本病例表现为右侧颈部Ⅱ区占位性病变,内部结构较为一致,边界较清,与周边无明显粘连,呈良性肿物的影像学表现,无特征性改变。3.3诊断与鉴别诊断由于本病发病部位不同,临床表现多样化,且临床症状无特异性,故术前诊断较困难,术前CT检查对确立本病有一定帮助,平扫显示病灶为密度均匀,边缘光整清楚或不光整软组织肿块,CT增强扫描,透明血管型UCD病灶强化程度稍稍低于或与邻近大血管同步明显强化,肿块明显强化是由于肿块内有较多血管和丰富毛细血管所致,而浆细胞UCD因血管成分较少呈不均匀轻中度强化,缺少特征表现;MCD其CT表现无明显肿块,往往表现一个或多个区域大小相似的淋巴结,增强呈轻中度强化。后两种情况,影像学表现不典型,术前诊断较困难。本病的早期确诊最终有赖于组织病理学检查。对于CD,不管临床或病理分类,都有组织学的共同特点:①具有完整的淋巴结基本结构;②淋巴滤泡和血管增生,在透明血管型滤泡、血管发生玻璃样变,滤泡生发中心萎缩;浆细胞型表现滤泡间质中有较多的浆细胞,滤泡生发中心增生。Frizzera提出CD的诊断标准:①UCD的诊断标准:单一部位淋巴结肿大,组织病理学上具有特征性增生,除外可能的其他原发病,多无全身症状及贫血、免疫球蛋白升高等(浆细胞型除外),肿物切除后长期存活;②MCD诊断标准:具有特征性组织病理学改变,显著淋巴结肿大累及多处外周淋巴结,多系统受累表现,排除其他病因。在鉴别诊断上,应与恶性淋巴瘤、淋巴结核、非特异性淋巴结炎等鉴别。与恶性淋巴瘤的鉴别包括以下几点:恶性淋巴瘤时淋巴结结构几乎完全被破坏,少有血管增生,具有单克隆的淋巴细胞或R-S细胞增生。对于常见的以淋巴结肿大为表现的疾病,如非特异性淋巴结炎及淋巴结核,两者鉴别要点在于:①体格检查上,前者多有按压痛而后者无明显按压痛;②血液生化检查可查及前者血液中中性粒细胞水平上升;③PPD试验鉴别。3.4治疗及预后基于既往的研究,CD应采取切除异常淋巴结的外科治疗方式。对于UCD,切除病变淋巴结是最为有效的方式。国外学者的临床追踪表明,UCD经过手术治疗后,基本没有复发倾向。值得一提的是,有全身症状的患者在切除病变淋巴结后,症状消失。但也有个别报道指出,外科手术治疗后9年,患者出现UCD复发。近年来的一个以278份病例报告为基础的META分析结果,得出:UCD外科手术治疗患者的死亡率为3.8%,而其他治疗方式为17.6%。因此,外科手段虽然十分有效,但也有失败复发的风险。无法完全切除的病变,剩余的部分将会进展,更多的此类病例则是保持或者无明显症状表现。利妥昔单抗是治疗MCD的药物,也尝试用于不能清除的UCD患者身上,并获得良好效果。至于MCD,因为受到检查方式的限制,外科切除不是主要的治疗方式。免疫治疗及化学疗法是MCD的基本治疗策略。MCD的预后评估也通常比UCD更为保守。因为现在还没有完整的MCD治疗方式的随机试验研究,治疗方式的选择评估受限于现有的病例报告。利妥昔单抗单独应用或者联合细胞抑制剂应用已经在许多患者身上取得了效果。利妥昔单抗的临床反应包括血清IL-6水平的下降以及其他HIV阳性患者病例中,随着HHV-8病毒水平下降而下降的细胞因子。在更严重的疾病情况下,出现了器官衰竭和体力下降的征象时,利妥昔单抗联合依托泊苷等化疗药物,可以提高5年生存率到90%。然而,化疗药物必须在组合方案中应用:单抗疗法联合长春花碱或者依托泊苷可以抑制MCD,但是一旦停用后,症状将会在数周内复发。最新的化疗方案,比如CHOP(cyclophosphamide,doxo-rubicin,vincristine,and prednisone),即环磷酰胺+多柔比星+长春新碱+**或者CVAD(cyclo-phosvnamide,pirarubicin,vincristine and dexameth-asone),即环磷酰胺+ 吡柔比星+长春新碱+**,在50%以上的患者中展示出持久的疗效。3.5相关疾病病程虽然CD是以淋巴结病变为主的疾病,但是无论UCD和MCD都有可能伴发着其他疾病发生。在法国的一系列病例中,8名UCD患者伴发恶性淋巴瘤,其中6位为B细胞非霍奇金淋巴瘤,2例为霍奇金淋巴瘤。8例患者中3例同时出现UCD和NHL的发病,即便前者与后者之间的平均诊断间隔为46个月,两者间的病理生理学关系尚未明了。除此之外,接近15% UCD患者发展出副肿瘤性天疱疮。MCD患者有相似的伴发疾病,而且比UCD患者有更多的出现多脏器功能紊乱的概率。例如,MCD患者有更高的恶性肿瘤的发病风险,比如卡波氏肉瘤和血液系统恶性肿瘤。13%MCD患者同时患有卡波氏肉瘤。NHL与MCD有显著的关系:15%到20%的患者伴发或者发展出NHL,预后较差,死亡率高达85%。HHV-联合HIV感染,被认为是MCD发展为NHL重要的风险因素。弥漫性大B细胞性淋巴瘤是HHV-阳性MCD患者最常见的淋巴瘤,这也是2008年WHO淋巴组织来源的恶性肿瘤分类中独特的一种。虽然潜在的机制尚未能确定,但霍奇金病可能与MCD 有关。MCD也伴随着POEMS 综合征,即多发性神经病变(polyneuropathy,P)、脏器肿大(organomegaly,O)、内分泌病变(endocrinopathy,E),单克隆γ球蛋白病(monoclonal gammopathy,M,也叫M-蛋白)和皮肤改变(skinchanges,S)。
来源:热娜古孜·阿不都米吉提,李世豪, 黄子贤,等. 颈部Castleman病临床报道暨文献回顾[J]. 口腔疾病防止, 2017, 25(2):110-114.
既往史
【讨论】
3.1发病概况及病因目前尚未有关于CD发病率的文献报道。文献报道国外患者平均年龄为35.6岁,国内为33.1岁。其发病率以女性多见,男∶女= 1∶4,但亦有文献报道发病率无性别差异。CD在临床上可以分为局灶型(Localized or unicentric CD,LCD orUCD)和多中心型(multicentric CD,MCD)。局灶型多发于青年,中位年龄约20岁,其中90%的局灶型病例为透明血管型,10%为浆细胞型。多中心型较局灶型少见,中位年龄57岁,男女发病率为1∶2。该病可发生在淋巴结存在的任何部位,国内报道以胸部的纵隔最多见(60%~70%),其次为颈部(10%~14%),腹部(5%~10%),腋部(2%~4%)等。国外报道局灶型好发部位为纵膈(30%),颈部(23%),腹部(20%),腹膜后腔(17%)及腋窝(5%),腹股沟(3%)和盆腔(2%)。既往对CD的病因研究认为,γ疱疹病毒HHV-8感染,血清白细胞介素6水平上升,VEGF引导的血管增生及滤泡树突状细胞不典型增生与CD的发生发展存在一定的相关性。3.2临床表现UCD患者常常没有自觉症状,于影像学检查时意外发现无痛性肿块。少部分患者可出现咳嗽,呼吸困难,或者其他全身症状。在大部分的UCD患者中表现为非进展性的临床表现。与此不同的是,MCD伴随全身炎症反应的系统表现:发热,盗汗,体重减轻,疲乏。MCD 患者常常有外周的淋巴结病,影响同一区域的多个淋巴结,实验室检测结果异常,比如:贫血、低蛋白血症、高丙种球蛋白血症和ESR、IL-6、IL-10水平升高。当以上的临床表现及实验室检查异常同时出现时,常常提示MCD的可能。影像学检查多表现为占位性病变或淋巴结肿大,无明显特征,发生于纵膈淋巴结的Castleman病在胸片上多表现为高密度团块影,边缘规整或有分叶,可单发或者多发,常见纵膈增宽的间接表现,累及肺实质。超声检查多表现为回声均匀的低回声团块影,包膜清晰完整;较大的病灶(大于5 cm)可表现为混杂不均匀的低回声,包膜不均匀。CT检查多表现为软组织肿块影,在动态扫描的变化过程类似于动脉。MRI 检查多表现为T1加权像上低信号和T2加权像上高信号影。MCD在影像学检查中还可见肝脾肿大及腹腔或胸腔积液表现。本病例表现为右侧颈部Ⅱ区占位性病变,内部结构较为一致,边界较清,与周边无明显粘连,呈良性肿物的影像学表现,无特征性改变。3.3诊断与鉴别诊断由于本病发病部位不同,临床表现多样化,且临床症状无特异性,故术前诊断较困难,术前CT检查对确立本病有一定帮助,平扫显示病灶为密度均匀,边缘光整清楚或不光整软组织肿块,CT增强扫描,透明血管型UCD病灶强化程度稍稍低于或与邻近大血管同步明显强化,肿块明显强化是由于肿块内有较多血管和丰富毛细血管所致,而浆细胞UCD因血管成分较少呈不均匀轻中度强化,缺少特征表现;MCD其CT表现无明显肿块,往往表现一个或多个区域大小相似的淋巴结,增强呈轻中度强化。后两种情况,影像学表现不典型,术前诊断较困难。本病的早期确诊最终有赖于组织病理学检查。对于CD,不管临床或病理分类,都有组织学的共同特点:①具有完整的淋巴结基本结构;②淋巴滤泡和血管增生,在透明血管型滤泡、血管发生玻璃样变,滤泡生发中心萎缩;浆细胞型表现滤泡间质中有较多的浆细胞,滤泡生发中心增生。Frizzera提出CD的诊断标准:①UCD的诊断标准:单一部位淋巴结肿大,组织病理学上具有特征性增生,除外可能的其他原发病,多无全身症状及贫血、免疫球蛋白升高等(浆细胞型除外),肿物切除后长期存活;②MCD诊断标准:具有特征性组织病理学改变,显著淋巴结肿大累及多处外周淋巴结,多系统受累表现,排除其他病因。在鉴别诊断上,应与恶性淋巴瘤、淋巴结核、非特异性淋巴结炎等鉴别。与恶性淋巴瘤的鉴别包括以下几点:恶性淋巴瘤时淋巴结结构几乎完全被破坏,少有血管增生,具有单克隆的淋巴细胞或R-S细胞增生。对于常见的以淋巴结肿大为表现的疾病,如非特异性淋巴结炎及淋巴结核,两者鉴别要点在于:①体格检查上,前者多有按压痛而后者无明显按压痛;②血液生化检查可查及前者血液中中性粒细胞水平上升;③PPD试验鉴别。3.4治疗及预后基于既往的研究,CD应采取切除异常淋巴结的外科治疗方式。对于UCD,切除病变淋巴结是最为有效的方式。国外学者的临床追踪表明,UCD经过手术治疗后,基本没有复发倾向。值得一提的是,有全身症状的患者在切除病变淋巴结后,症状消失。但也有个别报道指出,外科手术治疗后9年,患者出现UCD复发。近年来的一个以278份病例报告为基础的META分析结果,得出:UCD外科手术治疗患者的死亡率为3.8%,而其他治疗方式为17.6%。因此,外科手段虽然十分有效,但也有失败复发的风险。无法完全切除的病变,剩余的部分将会进展,更多的此类病例则是保持或者无明显症状表现。利妥昔单抗是治疗MCD的药物,也尝试用于不能清除的UCD患者身上,并获得良好效果。至于MCD,因为受到检查方式的限制,外科切除不是主要的治疗方式。免疫治疗及化学疗法是MCD的基本治疗策略。MCD的预后评估也通常比UCD更为保守。因为现在还没有完整的MCD治疗方式的随机试验研究,治疗方式的选择评估受限于现有的病例报告。利妥昔单抗单独应用或者联合细胞抑制剂应用已经在许多患者身上取得了效果。利妥昔单抗的临床反应包括血清IL-6水平的下降以及其他HIV阳性患者病例中,随着HHV-8病毒水平下降而下降的细胞因子。在更严重的疾病情况下,出现了器官衰竭和体力下降的征象时,利妥昔单抗联合依托泊苷等化疗药物,可以提高5年生存率到90%。然而,化疗药物必须在组合方案中应用:单抗疗法联合长春花碱或者依托泊苷可以抑制MCD,但是一旦停用后,症状将会在数周内复发。最新的化疗方案,比如CHOP(cyclophosphamide,doxo-rubicin,vincristine,and prednisone),即环磷酰胺+多柔比星+长春新碱+**或者CVAD(cyclo-phosvnamide,pirarubicin,vincristine and dexameth-asone),即环磷酰胺+ 吡柔比星+长春新碱+**,在50%以上的患者中展示出持久的疗效。3.5相关疾病病程虽然CD是以淋巴结病变为主的疾病,但是无论UCD和MCD都有可能伴发着其他疾病发生。在法国的一系列病例中,8名UCD患者伴发恶性淋巴瘤,其中6位为B细胞非霍奇金淋巴瘤,2例为霍奇金淋巴瘤。8例患者中3例同时出现UCD和NHL的发病,即便前者与后者之间的平均诊断间隔为46个月,两者间的病理生理学关系尚未明了。除此之外,接近15% UCD患者发展出副肿瘤性天疱疮。MCD患者有相似的伴发疾病,而且比UCD患者有更多的出现多脏器功能紊乱的概率。例如,MCD患者有更高的恶性肿瘤的发病风险,比如卡波氏肉瘤和血液系统恶性肿瘤。13%MCD患者同时患有卡波氏肉瘤。NHL与MCD有显著的关系:15%到20%的患者伴发或者发展出NHL,预后较差,死亡率高达85%。HHV-联合HIV感染,被认为是MCD发展为NHL重要的风险因素。弥漫性大B细胞性淋巴瘤是HHV-阳性MCD患者最常见的淋巴瘤,这也是2008年WHO淋巴组织来源的恶性肿瘤分类中独特的一种。虽然潜在的机制尚未能确定,但霍奇金病可能与MCD 有关。MCD也伴随着POEMS 综合征,即多发性神经病变(polyneuropathy,P)、脏器肿大(organomegaly,O)、内分泌病变(endocrinopathy,E),单克隆γ球蛋白病(monoclonal gammopathy,M,也叫M-蛋白)和皮肤改变(skinchanges,S)。
来源:热娜古孜·阿不都米吉提,李世豪, 黄子贤,等. 颈部Castleman病临床报道暨文献回顾[J]. 口腔疾病防止, 2017, 25(2):110-114.
查体
T:36.8℃,P:80次/分,R:18次/分,BP:120/80mmHg
【讨论】
3.1发病概况及病因目前尚未有关于CD发病率的文献报道。文献报道国外患者平均年龄为35.6岁,国内为33.1岁。其发病率以女性多见,男∶女= 1∶4,但亦有文献报道发病率无性别差异。CD在临床上可以分为局灶型(Localized or unicentric CD,LCD orUCD)和多中心型(multicentric CD,MCD)。局灶型多发于青年,中位年龄约20岁,其中90%的局灶型病例为透明血管型,10%为浆细胞型。多中心型较局灶型少见,中位年龄57岁,男女发病率为1∶2。该病可发生在淋巴结存在的任何部位,国内报道以胸部的纵隔最多见(60%~70%),其次为颈部(10%~14%),腹部(5%~10%),腋部(2%~4%)等。国外报道局灶型好发部位为纵膈(30%),颈部(23%),腹部(20%),腹膜后腔(17%)及腋窝(5%),腹股沟(3%)和盆腔(2%)。既往对CD的病因研究认为,γ疱疹病毒HHV-8感染,血清白细胞介素6水平上升,VEGF引导的血管增生及滤泡树突状细胞不典型增生与CD的发生发展存在一定的相关性。3.2临床表现UCD患者常常没有自觉症状,于影像学检查时意外发现无痛性肿块。少部分患者可出现咳嗽,呼吸困难,或者其他全身症状。在大部分的UCD患者中表现为非进展性的临床表现。与此不同的是,MCD伴随全身炎症反应的系统表现:发热,盗汗,体重减轻,疲乏。MCD 患者常常有外周的淋巴结病,影响同一区域的多个淋巴结,实验室检测结果异常,比如:贫血、低蛋白血症、高丙种球蛋白血症和ESR、IL-6、IL-10水平升高。当以上的临床表现及实验室检查异常同时出现时,常常提示MCD的可能。影像学检查多表现为占位性病变或淋巴结肿大,无明显特征,发生于纵膈淋巴结的Castleman病在胸片上多表现为高密度团块影,边缘规整或有分叶,可单发或者多发,常见纵膈增宽的间接表现,累及肺实质。超声检查多表现为回声均匀的低回声团块影,包膜清晰完整;较大的病灶(大于5 cm)可表现为混杂不均匀的低回声,包膜不均匀。CT检查多表现为软组织肿块影,在动态扫描的变化过程类似于动脉。MRI 检查多表现为T1加权像上低信号和T2加权像上高信号影。MCD在影像学检查中还可见肝脾肿大及腹腔或胸腔积液表现。本病例表现为右侧颈部Ⅱ区占位性病变,内部结构较为一致,边界较清,与周边无明显粘连,呈良性肿物的影像学表现,无特征性改变。3.3诊断与鉴别诊断由于本病发病部位不同,临床表现多样化,且临床症状无特异性,故术前诊断较困难,术前CT检查对确立本病有一定帮助,平扫显示病灶为密度均匀,边缘光整清楚或不光整软组织肿块,CT增强扫描,透明血管型UCD病灶强化程度稍稍低于或与邻近大血管同步明显强化,肿块明显强化是由于肿块内有较多血管和丰富毛细血管所致,而浆细胞UCD因血管成分较少呈不均匀轻中度强化,缺少特征表现;MCD其CT表现无明显肿块,往往表现一个或多个区域大小相似的淋巴结,增强呈轻中度强化。后两种情况,影像学表现不典型,术前诊断较困难。本病的早期确诊最终有赖于组织病理学检查。对于CD,不管临床或病理分类,都有组织学的共同特点:①具有完整的淋巴结基本结构;②淋巴滤泡和血管增生,在透明血管型滤泡、血管发生玻璃样变,滤泡生发中心萎缩;浆细胞型表现滤泡间质中有较多的浆细胞,滤泡生发中心增生。Frizzera提出CD的诊断标准:①UCD的诊断标准:单一部位淋巴结肿大,组织病理学上具有特征性增生,除外可能的其他原发病,多无全身症状及贫血、免疫球蛋白升高等(浆细胞型除外),肿物切除后长期存活;②MCD诊断标准:具有特征性组织病理学改变,显著淋巴结肿大累及多处外周淋巴结,多系统受累表现,排除其他病因。在鉴别诊断上,应与恶性淋巴瘤、淋巴结核、非特异性淋巴结炎等鉴别。与恶性淋巴瘤的鉴别包括以下几点:恶性淋巴瘤时淋巴结结构几乎完全被破坏,少有血管增生,具有单克隆的淋巴细胞或R-S细胞增生。对于常见的以淋巴结肿大为表现的疾病,如非特异性淋巴结炎及淋巴结核,两者鉴别要点在于:①体格检查上,前者多有按压痛而后者无明显按压痛;②血液生化检查可查及前者血液中中性粒细胞水平上升;③PPD试验鉴别。3.4治疗及预后基于既往的研究,CD应采取切除异常淋巴结的外科治疗方式。对于UCD,切除病变淋巴结是最为有效的方式。国外学者的临床追踪表明,UCD经过手术治疗后,基本没有复发倾向。值得一提的是,有全身症状的患者在切除病变淋巴结后,症状消失。但也有个别报道指出,外科手术治疗后9年,患者出现UCD复发。近年来的一个以278份病例报告为基础的META分析结果,得出:UCD外科手术治疗患者的死亡率为3.8%,而其他治疗方式为17.6%。因此,外科手段虽然十分有效,但也有失败复发的风险。无法完全切除的病变,剩余的部分将会进展,更多的此类病例则是保持或者无明显症状表现。利妥昔单抗是治疗MCD的药物,也尝试用于不能清除的UCD患者身上,并获得良好效果。至于MCD,因为受到检查方式的限制,外科切除不是主要的治疗方式。免疫治疗及化学疗法是MCD的基本治疗策略。MCD的预后评估也通常比UCD更为保守。因为现在还没有完整的MCD治疗方式的随机试验研究,治疗方式的选择评估受限于现有的病例报告。利妥昔单抗单独应用或者联合细胞抑制剂应用已经在许多患者身上取得了效果。利妥昔单抗的临床反应包括血清IL-6水平的下降以及其他HIV阳性患者病例中,随着HHV-8病毒水平下降而下降的细胞因子。在更严重的疾病情况下,出现了器官衰竭和体力下降的征象时,利妥昔单抗联合依托泊苷等化疗药物,可以提高5年生存率到90%。然而,化疗药物必须在组合方案中应用:单抗疗法联合长春花碱或者依托泊苷可以抑制MCD,但是一旦停用后,症状将会在数周内复发。最新的化疗方案,比如CHOP(cyclophosphamide,doxo-rubicin,vincristine,and prednisone),即环磷酰胺+多柔比星+长春新碱+**或者CVAD(cyclo-phosvnamide,pirarubicin,vincristine and dexameth-asone),即环磷酰胺+ 吡柔比星+长春新碱+**,在50%以上的患者中展示出持久的疗效。3.5相关疾病病程虽然CD是以淋巴结病变为主的疾病,但是无论UCD和MCD都有可能伴发着其他疾病发生。在法国的一系列病例中,8名UCD患者伴发恶性淋巴瘤,其中6位为B细胞非霍奇金淋巴瘤,2例为霍奇金淋巴瘤。8例患者中3例同时出现UCD和NHL的发病,即便前者与后者之间的平均诊断间隔为46个月,两者间的病理生理学关系尚未明了。除此之外,接近15% UCD患者发展出副肿瘤性天疱疮。MCD患者有相似的伴发疾病,而且比UCD患者有更多的出现多脏器功能紊乱的概率。例如,MCD患者有更高的恶性肿瘤的发病风险,比如卡波氏肉瘤和血液系统恶性肿瘤。13%MCD患者同时患有卡波氏肉瘤。NHL与MCD有显著的关系:15%到20%的患者伴发或者发展出NHL,预后较差,死亡率高达85%。HHV-联合HIV感染,被认为是MCD发展为NHL重要的风险因素。弥漫性大B细胞性淋巴瘤是HHV-阳性MCD患者最常见的淋巴瘤,这也是2008年WHO淋巴组织来源的恶性肿瘤分类中独特的一种。虽然潜在的机制尚未能确定,但霍奇金病可能与MCD 有关。MCD也伴随着POEMS 综合征,即多发性神经病变(polyneuropathy,P)、脏器肿大(organomegaly,O)、内分泌病变(endocrinopathy,E),单克隆γ球蛋白病(monoclonal gammopathy,M,也叫M-蛋白)和皮肤改变(skinchanges,S)。
来源:热娜古孜·阿不都米吉提,李世豪, 黄子贤,等. 颈部Castleman病临床报道暨文献回顾[J]. 口腔疾病防止, 2017, 25(2):110-114.
辅助检查
【讨论】
3.1发病概况及病因目前尚未有关于CD发病率的文献报道。文献报道国外患者平均年龄为35.6岁,国内为33.1岁。其发病率以女性多见,男∶女= 1∶4,但亦有文献报道发病率无性别差异。CD在临床上可以分为局灶型(Localized or unicentric CD,LCD orUCD)和多中心型(multicentric CD,MCD)。局灶型多发于青年,中位年龄约20岁,其中90%的局灶型病例为透明血管型,10%为浆细胞型。多中心型较局灶型少见,中位年龄57岁,男女发病率为1∶2。该病可发生在淋巴结存在的任何部位,国内报道以胸部的纵隔最多见(60%~70%),其次为颈部(10%~14%),腹部(5%~10%),腋部(2%~4%)等。国外报道局灶型好发部位为纵膈(30%),颈部(23%),腹部(20%),腹膜后腔(17%)及腋窝(5%),腹股沟(3%)和盆腔(2%)。既往对CD的病因研究认为,γ疱疹病毒HHV-8感染,血清白细胞介素6水平上升,VEGF引导的血管增生及滤泡树突状细胞不典型增生与CD的发生发展存在一定的相关性。3.2临床表现UCD患者常常没有自觉症状,于影像学检查时意外发现无痛性肿块。少部分患者可出现咳嗽,呼吸困难,或者其他全身症状。在大部分的UCD患者中表现为非进展性的临床表现。与此不同的是,MCD伴随全身炎症反应的系统表现:发热,盗汗,体重减轻,疲乏。MCD 患者常常有外周的淋巴结病,影响同一区域的多个淋巴结,实验室检测结果异常,比如:贫血、低蛋白血症、高丙种球蛋白血症和ESR、IL-6、IL-10水平升高。当以上的临床表现及实验室检查异常同时出现时,常常提示MCD的可能。影像学检查多表现为占位性病变或淋巴结肿大,无明显特征,发生于纵膈淋巴结的Castleman病在胸片上多表现为高密度团块影,边缘规整或有分叶,可单发或者多发,常见纵膈增宽的间接表现,累及肺实质。超声检查多表现为回声均匀的低回声团块影,包膜清晰完整;较大的病灶(大于5 cm)可表现为混杂不均匀的低回声,包膜不均匀。CT检查多表现为软组织肿块影,在动态扫描的变化过程类似于动脉。MRI 检查多表现为T1加权像上低信号和T2加权像上高信号影。MCD在影像学检查中还可见肝脾肿大及腹腔或胸腔积液表现。本病例表现为右侧颈部Ⅱ区占位性病变,内部结构较为一致,边界较清,与周边无明显粘连,呈良性肿物的影像学表现,无特征性改变。3.3诊断与鉴别诊断由于本病发病部位不同,临床表现多样化,且临床症状无特异性,故术前诊断较困难,术前CT检查对确立本病有一定帮助,平扫显示病灶为密度均匀,边缘光整清楚或不光整软组织肿块,CT增强扫描,透明血管型UCD病灶强化程度稍稍低于或与邻近大血管同步明显强化,肿块明显强化是由于肿块内有较多血管和丰富毛细血管所致,而浆细胞UCD因血管成分较少呈不均匀轻中度强化,缺少特征表现;MCD其CT表现无明显肿块,往往表现一个或多个区域大小相似的淋巴结,增强呈轻中度强化。后两种情况,影像学表现不典型,术前诊断较困难。本病的早期确诊最终有赖于组织病理学检查。对于CD,不管临床或病理分类,都有组织学的共同特点:①具有完整的淋巴结基本结构;②淋巴滤泡和血管增生,在透明血管型滤泡、血管发生玻璃样变,滤泡生发中心萎缩;浆细胞型表现滤泡间质中有较多的浆细胞,滤泡生发中心增生。Frizzera提出CD的诊断标准:①UCD的诊断标准:单一部位淋巴结肿大,组织病理学上具有特征性增生,除外可能的其他原发病,多无全身症状及贫血、免疫球蛋白升高等(浆细胞型除外),肿物切除后长期存活;②MCD诊断标准:具有特征性组织病理学改变,显著淋巴结肿大累及多处外周淋巴结,多系统受累表现,排除其他病因。在鉴别诊断上,应与恶性淋巴瘤、淋巴结核、非特异性淋巴结炎等鉴别。与恶性淋巴瘤的鉴别包括以下几点:恶性淋巴瘤时淋巴结结构几乎完全被破坏,少有血管增生,具有单克隆的淋巴细胞或R-S细胞增生。对于常见的以淋巴结肿大为表现的疾病,如非特异性淋巴结炎及淋巴结核,两者鉴别要点在于:①体格检查上,前者多有按压痛而后者无明显按压痛;②血液生化检查可查及前者血液中中性粒细胞水平上升;③PPD试验鉴别。3.4治疗及预后基于既往的研究,CD应采取切除异常淋巴结的外科治疗方式。对于UCD,切除病变淋巴结是最为有效的方式。国外学者的临床追踪表明,UCD经过手术治疗后,基本没有复发倾向。值得一提的是,有全身症状的患者在切除病变淋巴结后,症状消失。但也有个别报道指出,外科手术治疗后9年,患者出现UCD复发。近年来的一个以278份病例报告为基础的META分析结果,得出:UCD外科手术治疗患者的死亡率为3.8%,而其他治疗方式为17.6%。因此,外科手段虽然十分有效,但也有失败复发的风险。无法完全切除的病变,剩余的部分将会进展,更多的此类病例则是保持或者无明显症状表现。利妥昔单抗是治疗MCD的药物,也尝试用于不能清除的UCD患者身上,并获得良好效果。至于MCD,因为受到检查方式的限制,外科切除不是主要的治疗方式。免疫治疗及化学疗法是MCD的基本治疗策略。MCD的预后评估也通常比UCD更为保守。因为现在还没有完整的MCD治疗方式的随机试验研究,治疗方式的选择评估受限于现有的病例报告。利妥昔单抗单独应用或者联合细胞抑制剂应用已经在许多患者身上取得了效果。利妥昔单抗的临床反应包括血清IL-6水平的下降以及其他HIV阳性患者病例中,随着HHV-8病毒水平下降而下降的细胞因子。在更严重的疾病情况下,出现了器官衰竭和体力下降的征象时,利妥昔单抗联合依托泊苷等化疗药物,可以提高5年生存率到90%。然而,化疗药物必须在组合方案中应用:单抗疗法联合长春花碱或者依托泊苷可以抑制MCD,但是一旦停用后,症状将会在数周内复发。最新的化疗方案,比如CHOP(cyclophosphamide,doxo-rubicin,vincristine,and prednisone),即环磷酰胺+多柔比星+长春新碱+**或者CVAD(cyclo-phosvnamide,pirarubicin,vincristine and dexameth-asone),即环磷酰胺+ 吡柔比星+长春新碱+**,在50%以上的患者中展示出持久的疗效。3.5相关疾病病程虽然CD是以淋巴结病变为主的疾病,但是无论UCD和MCD都有可能伴发着其他疾病发生。在法国的一系列病例中,8名UCD患者伴发恶性淋巴瘤,其中6位为B细胞非霍奇金淋巴瘤,2例为霍奇金淋巴瘤。8例患者中3例同时出现UCD和NHL的发病,即便前者与后者之间的平均诊断间隔为46个月,两者间的病理生理学关系尚未明了。除此之外,接近15% UCD患者发展出副肿瘤性天疱疮。MCD患者有相似的伴发疾病,而且比UCD患者有更多的出现多脏器功能紊乱的概率。例如,MCD患者有更高的恶性肿瘤的发病风险,比如卡波氏肉瘤和血液系统恶性肿瘤。13%MCD患者同时患有卡波氏肉瘤。NHL与MCD有显著的关系:15%到20%的患者伴发或者发展出NHL,预后较差,死亡率高达85%。HHV-联合HIV感染,被认为是MCD发展为NHL重要的风险因素。弥漫性大B细胞性淋巴瘤是HHV-阳性MCD患者最常见的淋巴瘤,这也是2008年WHO淋巴组织来源的恶性肿瘤分类中独特的一种。虽然潜在的机制尚未能确定,但霍奇金病可能与MCD 有关。MCD也伴随着POEMS 综合征,即多发性神经病变(polyneuropathy,P)、脏器肿大(organomegaly,O)、内分泌病变(endocrinopathy,E),单克隆γ球蛋白病(monoclonal gammopathy,M,也叫M-蛋白)和皮肤改变(skinchanges,S)。
来源:热娜古孜·阿不都米吉提,李世豪, 黄子贤,等. 颈部Castleman病临床报道暨文献回顾[J]. 口腔疾病防止, 2017, 25(2):110-114.
【诊治过程】
初步诊断
【讨论】
3.1发病概况及病因目前尚未有关于CD发病率的文献报道。文献报道国外患者平均年龄为35.6岁,国内为33.1岁。其发病率以女性多见,男∶女= 1∶4,但亦有文献报道发病率无性别差异。CD在临床上可以分为局灶型(Localized or unicentric CD,LCD orUCD)和多中心型(multicentric CD,MCD)。局灶型多发于青年,中位年龄约20岁,其中90%的局灶型病例为透明血管型,10%为浆细胞型。多中心型较局灶型少见,中位年龄57岁,男女发病率为1∶2。该病可发生在淋巴结存在的任何部位,国内报道以胸部的纵隔最多见(60%~70%),其次为颈部(10%~14%),腹部(5%~10%),腋部(2%~4%)等。国外报道局灶型好发部位为纵膈(30%),颈部(23%),腹部(20%),腹膜后腔(17%)及腋窝(5%),腹股沟(3%)和盆腔(2%)。既往对CD的病因研究认为,γ疱疹病毒HHV-8感染,血清白细胞介素6水平上升,VEGF引导的血管增生及滤泡树突状细胞不典型增生与CD的发生发展存在一定的相关性。3.2临床表现UCD患者常常没有自觉症状,于影像学检查时意外发现无痛性肿块。少部分患者可出现咳嗽,呼吸困难,或者其他全身症状。在大部分的UCD患者中表现为非进展性的临床表现。与此不同的是,MCD伴随全身炎症反应的系统表现:发热,盗汗,体重减轻,疲乏。MCD 患者常常有外周的淋巴结病,影响同一区域的多个淋巴结,实验室检测结果异常,比如:贫血、低蛋白血症、高丙种球蛋白血症和ESR、IL-6、IL-10水平升高。当以上的临床表现及实验室检查异常同时出现时,常常提示MCD的可能。影像学检查多表现为占位性病变或淋巴结肿大,无明显特征,发生于纵膈淋巴结的Castleman病在胸片上多表现为高密度团块影,边缘规整或有分叶,可单发或者多发,常见纵膈增宽的间接表现,累及肺实质。超声检查多表现为回声均匀的低回声团块影,包膜清晰完整;较大的病灶(大于5 cm)可表现为混杂不均匀的低回声,包膜不均匀。CT检查多表现为软组织肿块影,在动态扫描的变化过程类似于动脉。MRI 检查多表现为T1加权像上低信号和T2加权像上高信号影。MCD在影像学检查中还可见肝脾肿大及腹腔或胸腔积液表现。本病例表现为右侧颈部Ⅱ区占位性病变,内部结构较为一致,边界较清,与周边无明显粘连,呈良性肿物的影像学表现,无特征性改变。3.3诊断与鉴别诊断由于本病发病部位不同,临床表现多样化,且临床症状无特异性,故术前诊断较困难,术前CT检查对确立本病有一定帮助,平扫显示病灶为密度均匀,边缘光整清楚或不光整软组织肿块,CT增强扫描,透明血管型UCD病灶强化程度稍稍低于或与邻近大血管同步明显强化,肿块明显强化是由于肿块内有较多血管和丰富毛细血管所致,而浆细胞UCD因血管成分较少呈不均匀轻中度强化,缺少特征表现;MCD其CT表现无明显肿块,往往表现一个或多个区域大小相似的淋巴结,增强呈轻中度强化。后两种情况,影像学表现不典型,术前诊断较困难。本病的早期确诊最终有赖于组织病理学检查。对于CD,不管临床或病理分类,都有组织学的共同特点:①具有完整的淋巴结基本结构;②淋巴滤泡和血管增生,在透明血管型滤泡、血管发生玻璃样变,滤泡生发中心萎缩;浆细胞型表现滤泡间质中有较多的浆细胞,滤泡生发中心增生。Frizzera提出CD的诊断标准:①UCD的诊断标准:单一部位淋巴结肿大,组织病理学上具有特征性增生,除外可能的其他原发病,多无全身症状及贫血、免疫球蛋白升高等(浆细胞型除外),肿物切除后长期存活;②MCD诊断标准:具有特征性组织病理学改变,显著淋巴结肿大累及多处外周淋巴结,多系统受累表现,排除其他病因。在鉴别诊断上,应与恶性淋巴瘤、淋巴结核、非特异性淋巴结炎等鉴别。与恶性淋巴瘤的鉴别包括以下几点:恶性淋巴瘤时淋巴结结构几乎完全被破坏,少有血管增生,具有单克隆的淋巴细胞或R-S细胞增生。对于常见的以淋巴结肿大为表现的疾病,如非特异性淋巴结炎及淋巴结核,两者鉴别要点在于:①体格检查上,前者多有按压痛而后者无明显按压痛;②血液生化检查可查及前者血液中中性粒细胞水平上升;③PPD试验鉴别。3.4治疗及预后基于既往的研究,CD应采取切除异常淋巴结的外科治疗方式。对于UCD,切除病变淋巴结是最为有效的方式。国外学者的临床追踪表明,UCD经过手术治疗后,基本没有复发倾向。值得一提的是,有全身症状的患者在切除病变淋巴结后,症状消失。但也有个别报道指出,外科手术治疗后9年,患者出现UCD复发。近年来的一个以278份病例报告为基础的META分析结果,得出:UCD外科手术治疗患者的死亡率为3.8%,而其他治疗方式为17.6%。因此,外科手段虽然十分有效,但也有失败复发的风险。无法完全切除的病变,剩余的部分将会进展,更多的此类病例则是保持或者无明显症状表现。利妥昔单抗是治疗MCD的药物,也尝试用于不能清除的UCD患者身上,并获得良好效果。至于MCD,因为受到检查方式的限制,外科切除不是主要的治疗方式。免疫治疗及化学疗法是MCD的基本治疗策略。MCD的预后评估也通常比UCD更为保守。因为现在还没有完整的MCD治疗方式的随机试验研究,治疗方式的选择评估受限于现有的病例报告。利妥昔单抗单独应用或者联合细胞抑制剂应用已经在许多患者身上取得了效果。利妥昔单抗的临床反应包括血清IL-6水平的下降以及其他HIV阳性患者病例中,随着HHV-8病毒水平下降而下降的细胞因子。在更严重的疾病情况下,出现了器官衰竭和体力下降的征象时,利妥昔单抗联合依托泊苷等化疗药物,可以提高5年生存率到90%。然而,化疗药物必须在组合方案中应用:单抗疗法联合长春花碱或者依托泊苷可以抑制MCD,但是一旦停用后,症状将会在数周内复发。最新的化疗方案,比如CHOP(cyclophosphamide,doxo-rubicin,vincristine,and prednisone),即环磷酰胺+多柔比星+长春新碱+**或者CVAD(cyclo-phosvnamide,pirarubicin,vincristine and dexameth-asone),即环磷酰胺+ 吡柔比星+长春新碱+**,在50%以上的患者中展示出持久的疗效。3.5相关疾病病程虽然CD是以淋巴结病变为主的疾病,但是无论UCD和MCD都有可能伴发着其他疾病发生。在法国的一系列病例中,8名UCD患者伴发恶性淋巴瘤,其中6位为B细胞非霍奇金淋巴瘤,2例为霍奇金淋巴瘤。8例患者中3例同时出现UCD和NHL的发病,即便前者与后者之间的平均诊断间隔为46个月,两者间的病理生理学关系尚未明了。除此之外,接近15% UCD患者发展出副肿瘤性天疱疮。MCD患者有相似的伴发疾病,而且比UCD患者有更多的出现多脏器功能紊乱的概率。例如,MCD患者有更高的恶性肿瘤的发病风险,比如卡波氏肉瘤和血液系统恶性肿瘤。13%MCD患者同时患有卡波氏肉瘤。NHL与MCD有显著的关系:15%到20%的患者伴发或者发展出NHL,预后较差,死亡率高达85%。HHV-联合HIV感染,被认为是MCD发展为NHL重要的风险因素。弥漫性大B细胞性淋巴瘤是HHV-阳性MCD患者最常见的淋巴瘤,这也是2008年WHO淋巴组织来源的恶性肿瘤分类中独特的一种。虽然潜在的机制尚未能确定,但霍奇金病可能与MCD 有关。MCD也伴随着POEMS 综合征,即多发性神经病变(polyneuropathy,P)、脏器肿大(organomegaly,O)、内分泌病变(endocrinopathy,E),单克隆γ球蛋白病(monoclonal gammopathy,M,也叫M-蛋白)和皮肤改变(skinchanges,S)。
来源:热娜古孜·阿不都米吉提,李世豪, 黄子贤,等. 颈部Castleman病临床报道暨文献回顾[J]. 口腔疾病防止, 2017, 25(2):110-114.
鉴别诊断
【讨论】
3.1发病概况及病因目前尚未有关于CD发病率的文献报道。文献报道国外患者平均年龄为35.6岁,国内为33.1岁。其发病率以女性多见,男∶女= 1∶4,但亦有文献报道发病率无性别差异。CD在临床上可以分为局灶型(Localized or unicentric CD,LCD orUCD)和多中心型(multicentric CD,MCD)。局灶型多发于青年,中位年龄约20岁,其中90%的局灶型病例为透明血管型,10%为浆细胞型。多中心型较局灶型少见,中位年龄57岁,男女发病率为1∶2。该病可发生在淋巴结存在的任何部位,国内报道以胸部的纵隔最多见(60%~70%),其次为颈部(10%~14%),腹部(5%~10%),腋部(2%~4%)等。国外报道局灶型好发部位为纵膈(30%),颈部(23%),腹部(20%),腹膜后腔(17%)及腋窝(5%),腹股沟(3%)和盆腔(2%)。既往对CD的病因研究认为,γ疱疹病毒HHV-8感染,血清白细胞介素6水平上升,VEGF引导的血管增生及滤泡树突状细胞不典型增生与CD的发生发展存在一定的相关性。3.2临床表现UCD患者常常没有自觉症状,于影像学检查时意外发现无痛性肿块。少部分患者可出现咳嗽,呼吸困难,或者其他全身症状。在大部分的UCD患者中表现为非进展性的临床表现。与此不同的是,MCD伴随全身炎症反应的系统表现:发热,盗汗,体重减轻,疲乏。MCD 患者常常有外周的淋巴结病,影响同一区域的多个淋巴结,实验室检测结果异常,比如:贫血、低蛋白血症、高丙种球蛋白血症和ESR、IL-6、IL-10水平升高。当以上的临床表现及实验室检查异常同时出现时,常常提示MCD的可能。影像学检查多表现为占位性病变或淋巴结肿大,无明显特征,发生于纵膈淋巴结的Castleman病在胸片上多表现为高密度团块影,边缘规整或有分叶,可单发或者多发,常见纵膈增宽的间接表现,累及肺实质。超声检查多表现为回声均匀的低回声团块影,包膜清晰完整;较大的病灶(大于5 cm)可表现为混杂不均匀的低回声,包膜不均匀。CT检查多表现为软组织肿块影,在动态扫描的变化过程类似于动脉。MRI 检查多表现为T1加权像上低信号和T2加权像上高信号影。MCD在影像学检查中还可见肝脾肿大及腹腔或胸腔积液表现。本病例表现为右侧颈部Ⅱ区占位性病变,内部结构较为一致,边界较清,与周边无明显粘连,呈良性肿物的影像学表现,无特征性改变。3.3诊断与鉴别诊断由于本病发病部位不同,临床表现多样化,且临床症状无特异性,故术前诊断较困难,术前CT检查对确立本病有一定帮助,平扫显示病灶为密度均匀,边缘光整清楚或不光整软组织肿块,CT增强扫描,透明血管型UCD病灶强化程度稍稍低于或与邻近大血管同步明显强化,肿块明显强化是由于肿块内有较多血管和丰富毛细血管所致,而浆细胞UCD因血管成分较少呈不均匀轻中度强化,缺少特征表现;MCD其CT表现无明显肿块,往往表现一个或多个区域大小相似的淋巴结,增强呈轻中度强化。后两种情况,影像学表现不典型,术前诊断较困难。本病的早期确诊最终有赖于组织病理学检查。对于CD,不管临床或病理分类,都有组织学的共同特点:①具有完整的淋巴结基本结构;②淋巴滤泡和血管增生,在透明血管型滤泡、血管发生玻璃样变,滤泡生发中心萎缩;浆细胞型表现滤泡间质中有较多的浆细胞,滤泡生发中心增生。Frizzera提出CD的诊断标准:①UCD的诊断标准:单一部位淋巴结肿大,组织病理学上具有特征性增生,除外可能的其他原发病,多无全身症状及贫血、免疫球蛋白升高等(浆细胞型除外),肿物切除后长期存活;②MCD诊断标准:具有特征性组织病理学改变,显著淋巴结肿大累及多处外周淋巴结,多系统受累表现,排除其他病因。在鉴别诊断上,应与恶性淋巴瘤、淋巴结核、非特异性淋巴结炎等鉴别。与恶性淋巴瘤的鉴别包括以下几点:恶性淋巴瘤时淋巴结结构几乎完全被破坏,少有血管增生,具有单克隆的淋巴细胞或R-S细胞增生。对于常见的以淋巴结肿大为表现的疾病,如非特异性淋巴结炎及淋巴结核,两者鉴别要点在于:①体格检查上,前者多有按压痛而后者无明显按压痛;②血液生化检查可查及前者血液中中性粒细胞水平上升;③PPD试验鉴别。3.4治疗及预后基于既往的研究,CD应采取切除异常淋巴结的外科治疗方式。对于UCD,切除病变淋巴结是最为有效的方式。国外学者的临床追踪表明,UCD经过手术治疗后,基本没有复发倾向。值得一提的是,有全身症状的患者在切除病变淋巴结后,症状消失。但也有个别报道指出,外科手术治疗后9年,患者出现UCD复发。近年来的一个以278份病例报告为基础的META分析结果,得出:UCD外科手术治疗患者的死亡率为3.8%,而其他治疗方式为17.6%。因此,外科手段虽然十分有效,但也有失败复发的风险。无法完全切除的病变,剩余的部分将会进展,更多的此类病例则是保持或者无明显症状表现。利妥昔单抗是治疗MCD的药物,也尝试用于不能清除的UCD患者身上,并获得良好效果。至于MCD,因为受到检查方式的限制,外科切除不是主要的治疗方式。免疫治疗及化学疗法是MCD的基本治疗策略。MCD的预后评估也通常比UCD更为保守。因为现在还没有完整的MCD治疗方式的随机试验研究,治疗方式的选择评估受限于现有的病例报告。利妥昔单抗单独应用或者联合细胞抑制剂应用已经在许多患者身上取得了效果。利妥昔单抗的临床反应包括血清IL-6水平的下降以及其他HIV阳性患者病例中,随着HHV-8病毒水平下降而下降的细胞因子。在更严重的疾病情况下,出现了器官衰竭和体力下降的征象时,利妥昔单抗联合依托泊苷等化疗药物,可以提高5年生存率到90%。然而,化疗药物必须在组合方案中应用:单抗疗法联合长春花碱或者依托泊苷可以抑制MCD,但是一旦停用后,症状将会在数周内复发。最新的化疗方案,比如CHOP(cyclophosphamide,doxo-rubicin,vincristine,and prednisone),即环磷酰胺+多柔比星+长春新碱+**或者CVAD(cyclo-phosvnamide,pirarubicin,vincristine and dexameth-asone),即环磷酰胺+ 吡柔比星+长春新碱+**,在50%以上的患者中展示出持久的疗效。3.5相关疾病病程虽然CD是以淋巴结病变为主的疾病,但是无论UCD和MCD都有可能伴发着其他疾病发生。在法国的一系列病例中,8名UCD患者伴发恶性淋巴瘤,其中6位为B细胞非霍奇金淋巴瘤,2例为霍奇金淋巴瘤。8例患者中3例同时出现UCD和NHL的发病,即便前者与后者之间的平均诊断间隔为46个月,两者间的病理生理学关系尚未明了。除此之外,接近15% UCD患者发展出副肿瘤性天疱疮。MCD患者有相似的伴发疾病,而且比UCD患者有更多的出现多脏器功能紊乱的概率。例如,MCD患者有更高的恶性肿瘤的发病风险,比如卡波氏肉瘤和血液系统恶性肿瘤。13%MCD患者同时患有卡波氏肉瘤。NHL与MCD有显著的关系:15%到20%的患者伴发或者发展出NHL,预后较差,死亡率高达85%。HHV-联合HIV感染,被认为是MCD发展为NHL重要的风险因素。弥漫性大B细胞性淋巴瘤是HHV-阳性MCD患者最常见的淋巴瘤,这也是2008年WHO淋巴组织来源的恶性肿瘤分类中独特的一种。虽然潜在的机制尚未能确定,但霍奇金病可能与MCD 有关。MCD也伴随着POEMS 综合征,即多发性神经病变(polyneuropathy,P)、脏器肿大(organomegaly,O)、内分泌病变(endocrinopathy,E),单克隆γ球蛋白病(monoclonal gammopathy,M,也叫M-蛋白)和皮肤改变(skinchanges,S)。
来源:热娜古孜·阿不都米吉提,李世豪, 黄子贤,等. 颈部Castleman病临床报道暨文献回顾[J]. 口腔疾病防止, 2017, 25(2):110-114.
诊断结果
【讨论】
3.1发病概况及病因目前尚未有关于CD发病率的文献报道。文献报道国外患者平均年龄为35.6岁,国内为33.1岁。其发病率以女性多见,男∶女= 1∶4,但亦有文献报道发病率无性别差异。CD在临床上可以分为局灶型(Localized or unicentric CD,LCD orUCD)和多中心型(multicentric CD,MCD)。局灶型多发于青年,中位年龄约20岁,其中90%的局灶型病例为透明血管型,10%为浆细胞型。多中心型较局灶型少见,中位年龄57岁,男女发病率为1∶2。该病可发生在淋巴结存在的任何部位,国内报道以胸部的纵隔最多见(60%~70%),其次为颈部(10%~14%),腹部(5%~10%),腋部(2%~4%)等。国外报道局灶型好发部位为纵膈(30%),颈部(23%),腹部(20%),腹膜后腔(17%)及腋窝(5%),腹股沟(3%)和盆腔(2%)。既往对CD的病因研究认为,γ疱疹病毒HHV-8感染,血清白细胞介素6水平上升,VEGF引导的血管增生及滤泡树突状细胞不典型增生与CD的发生发展存在一定的相关性。3.2临床表现UCD患者常常没有自觉症状,于影像学检查时意外发现无痛性肿块。少部分患者可出现咳嗽,呼吸困难,或者其他全身症状。在大部分的UCD患者中表现为非进展性的临床表现。与此不同的是,MCD伴随全身炎症反应的系统表现:发热,盗汗,体重减轻,疲乏。MCD 患者常常有外周的淋巴结病,影响同一区域的多个淋巴结,实验室检测结果异常,比如:贫血、低蛋白血症、高丙种球蛋白血症和ESR、IL-6、IL-10水平升高。当以上的临床表现及实验室检查异常同时出现时,常常提示MCD的可能。影像学检查多表现为占位性病变或淋巴结肿大,无明显特征,发生于纵膈淋巴结的Castleman病在胸片上多表现为高密度团块影,边缘规整或有分叶,可单发或者多发,常见纵膈增宽的间接表现,累及肺实质。超声检查多表现为回声均匀的低回声团块影,包膜清晰完整;较大的病灶(大于5 cm)可表现为混杂不均匀的低回声,包膜不均匀。CT检查多表现为软组织肿块影,在动态扫描的变化过程类似于动脉。MRI 检查多表现为T1加权像上低信号和T2加权像上高信号影。MCD在影像学检查中还可见肝脾肿大及腹腔或胸腔积液表现。本病例表现为右侧颈部Ⅱ区占位性病变,内部结构较为一致,边界较清,与周边无明显粘连,呈良性肿物的影像学表现,无特征性改变。3.3诊断与鉴别诊断由于本病发病部位不同,临床表现多样化,且临床症状无特异性,故术前诊断较困难,术前CT检查对确立本病有一定帮助,平扫显示病灶为密度均匀,边缘光整清楚或不光整软组织肿块,CT增强扫描,透明血管型UCD病灶强化程度稍稍低于或与邻近大血管同步明显强化,肿块明显强化是由于肿块内有较多血管和丰富毛细血管所致,而浆细胞UCD因血管成分较少呈不均匀轻中度强化,缺少特征表现;MCD其CT表现无明显肿块,往往表现一个或多个区域大小相似的淋巴结,增强呈轻中度强化。后两种情况,影像学表现不典型,术前诊断较困难。本病的早期确诊最终有赖于组织病理学检查。对于CD,不管临床或病理分类,都有组织学的共同特点:①具有完整的淋巴结基本结构;②淋巴滤泡和血管增生,在透明血管型滤泡、血管发生玻璃样变,滤泡生发中心萎缩;浆细胞型表现滤泡间质中有较多的浆细胞,滤泡生发中心增生。Frizzera提出CD的诊断标准:①UCD的诊断标准:单一部位淋巴结肿大,组织病理学上具有特征性增生,除外可能的其他原发病,多无全身症状及贫血、免疫球蛋白升高等(浆细胞型除外),肿物切除后长期存活;②MCD诊断标准:具有特征性组织病理学改变,显著淋巴结肿大累及多处外周淋巴结,多系统受累表现,排除其他病因。在鉴别诊断上,应与恶性淋巴瘤、淋巴结核、非特异性淋巴结炎等鉴别。与恶性淋巴瘤的鉴别包括以下几点:恶性淋巴瘤时淋巴结结构几乎完全被破坏,少有血管增生,具有单克隆的淋巴细胞或R-S细胞增生。对于常见的以淋巴结肿大为表现的疾病,如非特异性淋巴结炎及淋巴结核,两者鉴别要点在于:①体格检查上,前者多有按压痛而后者无明显按压痛;②血液生化检查可查及前者血液中中性粒细胞水平上升;③PPD试验鉴别。3.4治疗及预后基于既往的研究,CD应采取切除异常淋巴结的外科治疗方式。对于UCD,切除病变淋巴结是最为有效的方式。国外学者的临床追踪表明,UCD经过手术治疗后,基本没有复发倾向。值得一提的是,有全身症状的患者在切除病变淋巴结后,症状消失。但也有个别报道指出,外科手术治疗后9年,患者出现UCD复发。近年来的一个以278份病例报告为基础的META分析结果,得出:UCD外科手术治疗患者的死亡率为3.8%,而其他治疗方式为17.6%。因此,外科手段虽然十分有效,但也有失败复发的风险。无法完全切除的病变,剩余的部分将会进展,更多的此类病例则是保持或者无明显症状表现。利妥昔单抗是治疗MCD的药物,也尝试用于不能清除的UCD患者身上,并获得良好效果。至于MCD,因为受到检查方式的限制,外科切除不是主要的治疗方式。免疫治疗及化学疗法是MCD的基本治疗策略。MCD的预后评估也通常比UCD更为保守。因为现在还没有完整的MCD治疗方式的随机试验研究,治疗方式的选择评估受限于现有的病例报告。利妥昔单抗单独应用或者联合细胞抑制剂应用已经在许多患者身上取得了效果。利妥昔单抗的临床反应包括血清IL-6水平的下降以及其他HIV阳性患者病例中,随着HHV-8病毒水平下降而下降的细胞因子。在更严重的疾病情况下,出现了器官衰竭和体力下降的征象时,利妥昔单抗联合依托泊苷等化疗药物,可以提高5年生存率到90%。然而,化疗药物必须在组合方案中应用:单抗疗法联合长春花碱或者依托泊苷可以抑制MCD,但是一旦停用后,症状将会在数周内复发。最新的化疗方案,比如CHOP(cyclophosphamide,doxo-rubicin,vincristine,and prednisone),即环磷酰胺+多柔比星+长春新碱+**或者CVAD(cyclo-phosvnamide,pirarubicin,vincristine and dexameth-asone),即环磷酰胺+ 吡柔比星+长春新碱+**,在50%以上的患者中展示出持久的疗效。3.5相关疾病病程虽然CD是以淋巴结病变为主的疾病,但是无论UCD和MCD都有可能伴发着其他疾病发生。在法国的一系列病例中,8名UCD患者伴发恶性淋巴瘤,其中6位为B细胞非霍奇金淋巴瘤,2例为霍奇金淋巴瘤。8例患者中3例同时出现UCD和NHL的发病,即便前者与后者之间的平均诊断间隔为46个月,两者间的病理生理学关系尚未明了。除此之外,接近15% UCD患者发展出副肿瘤性天疱疮。MCD患者有相似的伴发疾病,而且比UCD患者有更多的出现多脏器功能紊乱的概率。例如,MCD患者有更高的恶性肿瘤的发病风险,比如卡波氏肉瘤和血液系统恶性肿瘤。13%MCD患者同时患有卡波氏肉瘤。NHL与MCD有显著的关系:15%到20%的患者伴发或者发展出NHL,预后较差,死亡率高达85%。HHV-联合HIV感染,被认为是MCD发展为NHL重要的风险因素。弥漫性大B细胞性淋巴瘤是HHV-阳性MCD患者最常见的淋巴瘤,这也是2008年WHO淋巴组织来源的恶性肿瘤分类中独特的一种。虽然潜在的机制尚未能确定,但霍奇金病可能与MCD 有关。MCD也伴随着POEMS 综合征,即多发性神经病变(polyneuropathy,P)、脏器肿大(organomegaly,O)、内分泌病变(endocrinopathy,E),单克隆γ球蛋白病(monoclonal gammopathy,M,也叫M-蛋白)和皮肤改变(skinchanges,S)。
来源:热娜古孜·阿不都米吉提,李世豪, 黄子贤,等. 颈部Castleman病临床报道暨文献回顾[J]. 口腔疾病防止, 2017, 25(2):110-114.
【其他】
【讨论】
3.1发病概况及病因目前尚未有关于CD发病率的文献报道。文献报道国外患者平均年龄为35.6岁,国内为33.1岁。其发病率以女性多见,男∶女= 1∶4,但亦有文献报道发病率无性别差异。CD在临床上可以分为局灶型(Localized or unicentric CD,LCD orUCD)和多中心型(multicentric CD,MCD)。局灶型多发于青年,中位年龄约20岁,其中90%的局灶型病例为透明血管型,10%为浆细胞型。多中心型较局灶型少见,中位年龄57岁,男女发病率为1∶2。该病可发生在淋巴结存在的任何部位,国内报道以胸部的纵隔最多见(60%~70%),其次为颈部(10%~14%),腹部(5%~10%),腋部(2%~4%)等。国外报道局灶型好发部位为纵膈(30%),颈部(23%),腹部(20%),腹膜后腔(17%)及腋窝(5%),腹股沟(3%)和盆腔(2%)。既往对CD的病因研究认为,γ疱疹病毒HHV-8感染,血清白细胞介素6水平上升,VEGF引导的血管增生及滤泡树突状细胞不典型增生与CD的发生发展存在一定的相关性。3.2临床表现UCD患者常常没有自觉症状,于影像学检查时意外发现无痛性肿块。少部分患者可出现咳嗽,呼吸困难,或者其他全身症状。在大部分的UCD患者中表现为非进展性的临床表现。与此不同的是,MCD伴随全身炎症反应的系统表现:发热,盗汗,体重减轻,疲乏。MCD 患者常常有外周的淋巴结病,影响同一区域的多个淋巴结,实验室检测结果异常,比如:贫血、低蛋白血症、高丙种球蛋白血症和ESR、IL-6、IL-10水平升高。当以上的临床表现及实验室检查异常同时出现时,常常提示MCD的可能。影像学检查多表现为占位性病变或淋巴结肿大,无明显特征,发生于纵膈淋巴结的Castleman病在胸片上多表现为高密度团块影,边缘规整或有分叶,可单发或者多发,常见纵膈增宽的间接表现,累及肺实质。超声检查多表现为回声均匀的低回声团块影,包膜清晰完整;较大的病灶(大于5 cm)可表现为混杂不均匀的低回声,包膜不均匀。CT检查多表现为软组织肿块影,在动态扫描的变化过程类似于动脉。MRI 检查多表现为T1加权像上低信号和T2加权像上高信号影。MCD在影像学检查中还可见肝脾肿大及腹腔或胸腔积液表现。本病例表现为右侧颈部Ⅱ区占位性病变,内部结构较为一致,边界较清,与周边无明显粘连,呈良性肿物的影像学表现,无特征性改变。3.3诊断与鉴别诊断由于本病发病部位不同,临床表现多样化,且临床症状无特异性,故术前诊断较困难,术前CT检查对确立本病有一定帮助,平扫显示病灶为密度均匀,边缘光整清楚或不光整软组织肿块,CT增强扫描,透明血管型UCD病灶强化程度稍稍低于或与邻近大血管同步明显强化,肿块明显强化是由于肿块内有较多血管和丰富毛细血管所致,而浆细胞UCD因血管成分较少呈不均匀轻中度强化,缺少特征表现;MCD其CT表现无明显肿块,往往表现一个或多个区域大小相似的淋巴结,增强呈轻中度强化。后两种情况,影像学表现不典型,术前诊断较困难。本病的早期确诊最终有赖于组织病理学检查。对于CD,不管临床或病理分类,都有组织学的共同特点:①具有完整的淋巴结基本结构;②淋巴滤泡和血管增生,在透明血管型滤泡、血管发生玻璃样变,滤泡生发中心萎缩;浆细胞型表现滤泡间质中有较多的浆细胞,滤泡生发中心增生。Frizzera提出CD的诊断标准:①UCD的诊断标准:单一部位淋巴结肿大,组织病理学上具有特征性增生,除外可能的其他原发病,多无全身症状及贫血、免疫球蛋白升高等(浆细胞型除外),肿物切除后长期存活;②MCD诊断标准:具有特征性组织病理学改变,显著淋巴结肿大累及多处外周淋巴结,多系统受累表现,排除其他病因。在鉴别诊断上,应与恶性淋巴瘤、淋巴结核、非特异性淋巴结炎等鉴别。与恶性淋巴瘤的鉴别包括以下几点:恶性淋巴瘤时淋巴结结构几乎完全被破坏,少有血管增生,具有单克隆的淋巴细胞或R-S细胞增生。对于常见的以淋巴结肿大为表现的疾病,如非特异性淋巴结炎及淋巴结核,两者鉴别要点在于:①体格检查上,前者多有按压痛而后者无明显按压痛;②血液生化检查可查及前者血液中中性粒细胞水平上升;③PPD试验鉴别。3.4治疗及预后基于既往的研究,CD应采取切除异常淋巴结的外科治疗方式。对于UCD,切除病变淋巴结是最为有效的方式。国外学者的临床追踪表明,UCD经过手术治疗后,基本没有复发倾向。值得一提的是,有全身症状的患者在切除病变淋巴结后,症状消失。但也有个别报道指出,外科手术治疗后9年,患者出现UCD复发。近年来的一个以278份病例报告为基础的META分析结果,得出:UCD外科手术治疗患者的死亡率为3.8%,而其他治疗方式为17.6%。因此,外科手段虽然十分有效,但也有失败复发的风险。无法完全切除的病变,剩余的部分将会进展,更多的此类病例则是保持或者无明显症状表现。利妥昔单抗是治疗MCD的药物,也尝试用于不能清除的UCD患者身上,并获得良好效果。至于MCD,因为受到检查方式的限制,外科切除不是主要的治疗方式。免疫治疗及化学疗法是MCD的基本治疗策略。MCD的预后评估也通常比UCD更为保守。因为现在还没有完整的MCD治疗方式的随机试验研究,治疗方式的选择评估受限于现有的病例报告。利妥昔单抗单独应用或者联合细胞抑制剂应用已经在许多患者身上取得了效果。利妥昔单抗的临床反应包括血清IL-6水平的下降以及其他HIV阳性患者病例中,随着HHV-8病毒水平下降而下降的细胞因子。在更严重的疾病情况下,出现了器官衰竭和体力下降的征象时,利妥昔单抗联合依托泊苷等化疗药物,可以提高5年生存率到90%。然而,化疗药物必须在组合方案中应用:单抗疗法联合长春花碱或者依托泊苷可以抑制MCD,但是一旦停用后,症状将会在数周内复发。最新的化疗方案,比如CHOP(cyclophosphamide,doxo-rubicin,vincristine,and prednisone),即环磷酰胺+多柔比星+长春新碱+**或者CVAD(cyclo-phosvnamide,pirarubicin,vincristine and dexameth-asone),即环磷酰胺+ 吡柔比星+长春新碱+**,在50%以上的患者中展示出持久的疗效。3.5相关疾病病程虽然CD是以淋巴结病变为主的疾病,但是无论UCD和MCD都有可能伴发着其他疾病发生。在法国的一系列病例中,8名UCD患者伴发恶性淋巴瘤,其中6位为B细胞非霍奇金淋巴瘤,2例为霍奇金淋巴瘤。8例患者中3例同时出现UCD和NHL的发病,即便前者与后者之间的平均诊断间隔为46个月,两者间的病理生理学关系尚未明了。除此之外,接近15% UCD患者发展出副肿瘤性天疱疮。MCD患者有相似的伴发疾病,而且比UCD患者有更多的出现多脏器功能紊乱的概率。例如,MCD患者有更高的恶性肿瘤的发病风险,比如卡波氏肉瘤和血液系统恶性肿瘤。13%MCD患者同时患有卡波氏肉瘤。NHL与MCD有显著的关系:15%到20%的患者伴发或者发展出NHL,预后较差,死亡率高达85%。HHV-联合HIV感染,被认为是MCD发展为NHL重要的风险因素。弥漫性大B细胞性淋巴瘤是HHV-阳性MCD患者最常见的淋巴瘤,这也是2008年WHO淋巴组织来源的恶性肿瘤分类中独特的一种。虽然潜在的机制尚未能确定,但霍奇金病可能与MCD 有关。MCD也伴随着POEMS 综合征,即多发性神经病变(polyneuropathy,P)、脏器肿大(organomegaly,O)、内分泌病变(endocrinopathy,E),单克隆γ球蛋白病(monoclonal gammopathy,M,也叫M-蛋白)和皮肤改变(skinchanges,S)。
来源:热娜古孜·阿不都米吉提,李世豪, 黄子贤,等. 颈部Castleman病临床报道暨文献回顾[J]. 口腔疾病防止, 2017, 25(2):110-114.
病例来源:爱爱医
版权声明:站所注明来源为"爱爱医"的文章,版权归作者与本站共同所有,非经授权不得转载。本站所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,如果您认为我们的转载侵犯了您的权益,请及时通过电话(400-626-9910)或邮箱(zlzs@120.net)通知我们,我们将第一时间处理,感谢。







全部评论